the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Jun 2026
| 22 Jun 2026
Dehydration reactions in guano-derived minerals: the taranakite-to-francoanellite transformation
Nicola Campomenosi
Fabio Bellatreccia
Donato Belmonte
Jo De Waele
Cristina Carbone
Bat guano deposits in caves host diverse assemblages of authigenic phosphates and sulfates. Previous field observations have proposed that some of them (e.g. brushite, gypsum, taranakite) can be subject to dehydration reactions as a response to varying environmental conditions. To evaluate the thermodynamic and kinetic constrains that regulate the dehydration reaction leading guano-derived taranakite (K3Al5(PO3OH)6(PO4)2 ⋅ 18H2O) to transform into francoanellite (K3Al5(PO3OH)6(PO4)2 ⋅ 12H2O), we conducted a multi-analytical in situ investigation consisting of temperature-resolved X-ray powder diffraction, Fourier transform infrared, and micro-Raman spectroscopy on a sample of taranakite. Thermodynamic calculations were also performed by means of the polyhedral approach to predict the phase transformation temperature at equilibrium, estimated to be 369.12 K (95.97 °C). Laboratory experiments conducted under increasing temperature conditions confirmed that the breaking of weak hydrogen bonds between interlayer water molecules and the aluminophosphate layers of taranakite is responsible for the onset of the dehydration reaction. Monitoring the evolution of the phosphate-stretching Raman peaks over time under isothermal conditions enabled us to estimate the activation energy of the process to be 7.6 ± 0.7 kJ mol−1 by means of the “time to a given fraction” method. The release of heat concomitant to the oxidation of organic matter in guano-admixed cave sediments is proposed as a viable trigger for the transformation, given the low kinetic barrier associated with it.
- Article
(5435 KB) - Full-text XML
-
Supplement
(480 KB) - BibTeX
- EndNote




Phosphate minerals are found in a wide variety of environments, spanning meteorites and planetary bodies (Postberg et al., 2023; Wu et al., 2024) to soils (Rothe et al., 2016). The most volumetrically abundant phosphate deposits are represented by sedimentary phosphorites, mainly consisting of apatite group minerals (Filippelli, 2011), while highly fractionated granitic pegmatites and the parageneses resulting from their secondary modifications bear the highest diversity in terms of structurally complex phosphates (Hawthorne, 1998). In the context of mineral evolution, phosphates are also the most prominent exponents of the recent (<0.4 Ga) and peculiar paragenetic mode of guano- and urine-derived minerals (Hazen and Morrison, 2022; Paragenetic Mode 52 (Mindat), 2025). Their occurrence is particularly favoured in cave environments, where large amounts of bat guano can accumulate over time due to the steady presence of bat colonies. Consequently, the organic matter constituting guano is microbially processed and oxidised, causing the release of phosphoric acid and the alteration and dissolution of carbonate bedrocks and siliciclastic sediments (Dandurand et al., 2019; Onac, 2019). This leads to important modifications in the mineralogical assemblages through massive precipitation of phosphate and sulfate minerals (Onac et al., 2002; Audra et al., 2021). The presence of bat colonies and the degradation of guano also cause perturbations in the cave atmosphere through the release of CO2 and NH3 and local increases in temperature and relative humidity (RH) levels. These conditions promote condensation–corrosion processes and lead to intense dissolution of cave walls and consequent enlargement of cave passages (Lundberg and McFarlane, 2012; Barriquand et al., 2021; Langford et al., 2025). Therefore, the presence of phosphate minerals and of biocorrosion-derived morphologies has recently become a fundamental proxy for the reconstruction of post-speleogenetic evolution processes (Farrant et al., 2025), such as the post-depositional transformation of cave sediments (Rellini et al., 2013) and the dissolution or alteration of archeological remains (i.e. stone tools and bones) (Sokol et al., 2022).
Although the importance of authigenic phosphates in cave deposits has been widely recognised, few studies have focused on the transformations they are subdued to in response to local changes in the micro-environments wherein they occur (e.g. in deposits of bat guano). Dumitraş and Marincea (2021) investigated the phosphate–sulfate assemblage derived from bat guano degradation in Gura Dobrogei Cave (Romania). The authors distinguished a set of “primary” phosphates and sulfates, namely hydroxylapatite, brushite, ardealite, gypsum, and taranakite, directly precipitating due to interactions between guano and the cave bedrock or sediments and a set of “secondary” phases, deriving from the partial dehydration of the former, presumably at temperatures of up to 373 K. In the latter group, they identified monetite, bassanite, and francoanellite, replacing brushite, gypsum, and taranakite, respectively, by means of topotactic substitution. They pointed to exothermic reactions regulating the oxidation of organic matter and ammonia present in guano mounds and in sediments admixed with guano as the most probable sources of heat necessary to trigger the onset of the dehydration reactions. A previous mineralogical study conducted on the products of guano degradation in Măgurici Cave (Romania) by Onac and Vereş (2003) also lead to the identification of monetite, bassanite, and francoanellite; in this case, however, the authors argued that their formation could be explained by low-RH conditions without including thermal processes in the discussion. Descriptive studies conducted by combining field and laboratory work are certainly fundamental to define the relations between secondary mineral assemblages and their genetic environments. Nonetheless, more analytical work under ambient and non-ambient conditions is needed to better evaluate the thermal behaviour and stability conditions of secondary phosphate minerals. In this context, we chose to investigate the transformation of taranakite into francoanellite by dehydration.
Taranakite (K3Al5(PO3OH)6(PO4)2 ⋅ 18H2O, space group ) is one of the most commonly occurring phases in phosphatised cave sediments. This mineral was first documented by Hector and Skey (1865) as an alteration product of trachytic rocks by seabird guano on the Sugar Loaf Islands (New Zealand) and has since then been reported from a variety of caves, always associated with the presence of recent or subfossil bat guano (Murray and Dietrich, 1956; Balenzano et al., 1974; Sakae and Sudo, 1975; Shahack-Gross et al., 2004; Sauro et al., 2014; Tămaş et al., 2014; Queffelec et al., 2018; Audra et al., 2019; Sokol et al., 2022). Taranakite lattice constants, thermal stability, and water and minor ammonium content were determined by Smith and Brown (1959) and Arlidge et al. (1963). The crystal structure was solved much later by Dick et al. (1998) on synthetic analogues of taranakite. The mineral has a layered structure; within a unit cell, six K3Al5(PO3OH)6(PO4)2 ⋅ 12H2O layers are interstratified with interlayers containing six H2O molecules (Fig. 1a), where the latter are arranged in a hexagonal geometry closely resembling the one in the structure of ice Ih (Peterson and Levy, 1957) and of fibroferrite (Ventruti et al., 2016). A network of hydrogen bonds (HBs) sustains the structure and can be subdivided in HBs internal to the layers; HBs inside the interlayers, holding together interstitial hexagonal rings of water molecules; and HBs between interlayers and layers, serving as bridges between interstitial water molecules and phosphate – coordinating oxygen or structural water molecules which form part of the Al coordination octahedra (Dick et al., 1998).
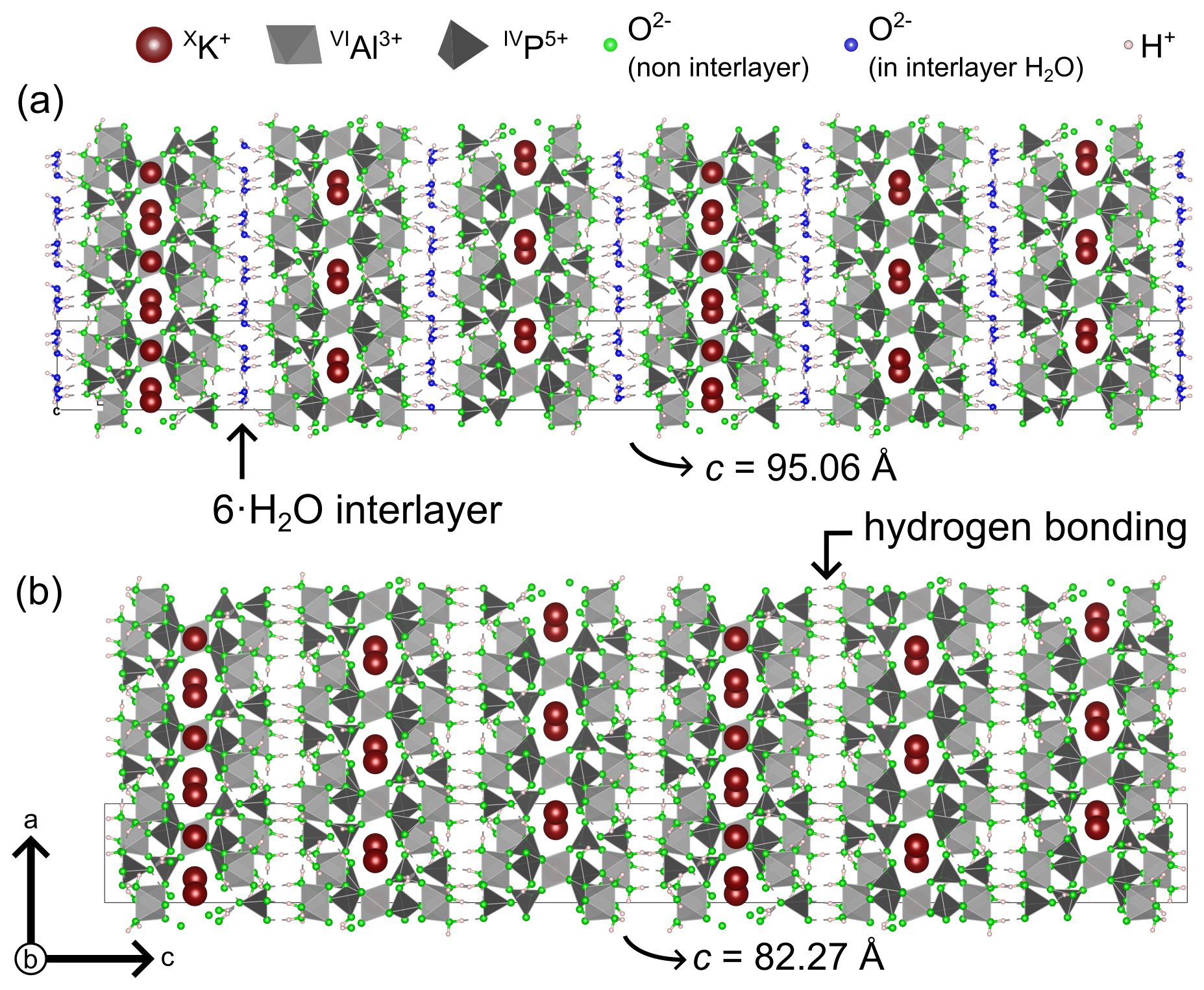
Figure 1The structures of taranakite (a) and francoanellite (b), as seen along the b axis. The thin solid dark lines delimitate the unit cells. Generated with VESTA (Momma and Izumi, 2008).
Upon heating, taranakite is subject to partial dehydration, which leads to the formation of francoanellite (K3Al5(PO3OH)6(PO4)2 ⋅ 12H2O, space group ), a phase which was first observed in the caves of Castellana (Apulia, Italy) by Balenzano et al. (1976). Other documented occurrences of francoanellite in nature are substantially limited (Balenzano et al., 1979; Chiorboli, 1984; Cancian, 1984; Dell'Anna et al., 1989; Hill and Forti, 1997, and references therein; Onac and Vereş, 2003; Dumitraş and Marincea, 2021) but are all related to cave environments. Given the rarity of this mineral, it remains difficult to assess whether its formation due to taranakite partial dehydration is a rare or widespread phenomenon. The complexity of such environments represents a further obstacle to the investigation of its conditions of formation.
The structure of a synthetic francoanellite, solved by Dick and Zeiske (1998), closely resembles the one of taranakite but lacks the water interlayers, which are replaced by HBs connecting P-coordinating oxygen to Al octahedra (Fig. 1b). The temperature conditions that induce this phase transformation have been the subject of numerous investigations by means of thermogravimetry differential thermal analysis (TG-DTA) and X-ray powder diffraction (XRPD) on the products of thermal treatment (Arlidge et al., 1963; Balenzano et al., 1974; Sakae and Sudo, 1975; Marincea and Dumitraş, 2003), which resulted in reports for the onset of the dehydration reaction spanning between 368 and 403 K. Nonetheless, a more precise estimate of the temperature of transformation from in situ experiments is still lacking, and the mechanisms that control the dehydration (i.e. the release of the water interlayers from the structure) remain unaddressed.
Intramineral processes (e.g. dehydration reactions), on par with intermineral reactions, define the mineral assemblages we observe in nature (Putnis, 1992). The complexity of mineral structures (Krivovichev et al., 2022) and the variety of mineralogical assemblages (Hazen and Morrison, 2022) increase dramatically in low-temperature, wet environments. This is particularly true in, but not restricted to, cave environments, where factors such as air-circulation-driven condensation–corrosion (Badino et al., 2011; Onac and Forti, 2011; Audra et al., 2026), changes in hydrological regime (Piccini et al., 2021), microbial activity, and biogeochemical reactions (Forti, 2001; Lazaridis et al., 2024; Bernardini et al., 2021) are intricately involved in the genesis and transformation of minerals. A detailed description of the mechanisms and energetics which regulate phase transformations is fundamental to better constrain how the complex interplay of multiple natural processes affects the stability and properties of secondary minerals. Such investigations are also relevant to extraterrestrial environments such as the Martian regolith, where the presence of hydrated sulfates (Siljeström et al., 2024) and Fe phosphates (Kizovski et al., 2025) has been confirmed and might help future reconstructions of the hydrologic and atmospheric evolution of Mars (Pineau et al., 2025, and references therein). A way to gain a more comprehensive understanding of their dehydration dynamics is to combine high-temperature (HT) XRPD, which detects the effects of the dehydration process in the long-range order, with HT Fourier transform infrared (FTIR) and Raman spectroscopy, allowing us to directly investigate changes in the hydration state and behaviour of water molecules within mineral structures (e.g. Putnis et al., 1990). In situ HT spectroscopic methods have been successfully applied to study the dehydration kinetics of gypsum and goethite (Prasad et al., 2001; Sendova et al., 2017). Unlike gypsum, Fe oxyhydroxides, and clay minerals, the mechanisms of dehydration of which have been the subject of a variety of studies implementing different theoretical and experimental approaches, hydrated secondary phosphate minerals are seldom considered in thermodynamic and kinetic studies. The aim of this work is to give a more comprehensive characterisation of the temperature conditions that determine the onset of the taranakite-to-francoanellite dehydration reaction and to estimate its empirical activation energy. For this purpose, we use a multimethodological experimental approach consisting of in situ HT XRPD, FTIR, and micro-Raman spectroscopy combined with equilibrium thermodynamic calculations.
2.1 Sample provenance and previous characterisation
The taranakite sample was collected in the Pollera Cave (Liguria, Italy) at the contact between a subfossil bat guano mound and a siliciclastic deposit of alluvial origin. A preliminary characterisation via XRPD and scanning electron microscopy (SEM), followed by TG-DTA experiments coupled with IR analysis of the exhaust gases, as conducted by Casale et al. (2025), allowed us to determine the mass losses and associated water release in the temperature range of 293–1473 K and to confirm the presence of a low amount of NH in the mineral, substituting for K+. The NH ↔ K+ substitution in synthetic taranakite had previously been documented by Arlidge et al. (1963), and, more recently, a natural NH analogue of taranakite, with the formula (NH4)3Al5(PO3OH)6(PO4)2 ⋅ 18H2O, was discovered in a sample from the Skipton Caves (Australia) and was accepted as a new mineral by the International Mineralogical Association, with the name macivorite (Elliott et al., 2025).
2.2 Thermodynamic calculations
A requirement for the thermodynamic calculation of the equilibrium temperature of the taranakite-to-francoanellite dehydration reaction is the knowledge of the Gibbs free energy of formation (Gf) under varying temperature conditions of the two minerals and of the water vapour liberated during the transformation. In order to obtain the thermochemical values of the crystalline phosphates, we adopted the polyhedral approximation (Hazen, 1985, 1988), which allows us to derive the bulk properties of minerals via the summation of the individual contributions of cation coordination polyhedra in the structure. Chermak and Rimstidt (1989) demonstrated that the polyhedral approach can be applied to derive the bulk thermodynamic properties of silicates. The polyhedral contributions can also be calculated under varying temperature conditions through the derivation of appropriate temperature functions (Chermak and Rimstidt, 1990). The values of polyhedral contributions to the Gibbs free energy of formation (gi) were derived by La Iglesia (2009) through least-squares regression of experimental thermochemical parameters from a set of 31 selected phosphates. The inferred thermodynamic properties proved to be accurate, with deviations of less than 2 % with respect to the experimental values of an independent set of 18 minerals which were not used for the regression. The same author also extended the calculation to high temperatures and published the temperature functions of the individual contributions of 16 basic polyhedral units, demonstrating their soundness by comparing the Gibbs free energies of formation calculated for well-characterised phosphates (i.e. berlinite, whitlockite, fluorapatite, and hydroxylapatite) in the range of 400–700 K, with thermodynamic tabulations by Robie et al. (1978). The Gibbs free energy of formation of H2O(g) was obtained by polynomial interpolation of the thermodynamic data tabulated by Barin (1995). The obtained set of temperature-dependent thermodynamic parameters allows us to easily calculate the temperature of equilibrium of the target reaction, as shown in Sect. 3.1.1.
2.3 HT in situ powder X-ray diffraction (XRPD)
XRPD analysis was performed at the Department of Geosciences of the University of Padua using a Philips X'Pert Pro diffractometer equipped with a long-fine-focus Co-anode tube (Co Kα=1.78901 Å) working at 40 kV–40 mA, a 240 mm goniometer radius operating in θ–θ geometry, and a Bragg–BrentanoHD (BBHD) module. Incident beam optics included a fixed 0.36° divergence slit and 0.4 rad Soller slits. The diffracted beam optics included an anti-scatter slit of 5 mm aperture, 0.04 rad Soller slits, and an X'Celerator detector with a 2.122° 2θ active length. For the acquisition of a preliminary scan at room temperature (RT), the powder sample was placed on a low-background Si sample holder. The acquisition was performed in 0.033° 2θ steps, with a measuring time of 1 s per step, in the angular range of 3–85° 2θ. The HT XRPD experiments were carried out in static air using an Anton Paar high-temperature cell (HTK16), increasing the temperature in steps of 10–20 K with a heating rate of 10 K min−1 from RT (298 K) to 438 K. The powder sample was placed on top of a Pt foil mounted on top of the Pt heating strip in the high-temperature chamber. The temperature was monitored using a Pt10 % Rh–Pt thermocouple. At every step, the temperature was kept constant, and three scans lasting 22 min each were performed in continuous mode with 0.033° 2θ steps, with a measuring time of 1 s per step, in the angular range of 4–48° 2θ. The lattice parameters were derived through Rietveld refinements of the XRPD data using the Profex software (Döbelin and Kleeberg, 2015) based on the BGMN programme. The structure files of taranakite and francoanellite were built from the CIF files available on the American Mineralogist Crystal Structure Database (AMCSD). Peak positions were refined with parameters accounting for sample displacement and cell dimensions, while peak broadening was modelled using isotropic microstrain and crystallite size parameters. As data collection was performed in reflection geometry on front-loaded powders, a fourth-order spherical harmonics function was used to correct for preferred orientation.
2.4 HT in situ Fourier transform infrared (FTIR) spectroscopy
FTIR analysis was performed at the Department of Science, Roma Tre University, using a Nicolet iS50 FTIR spectrometer equipped with a DTGS detector and a KBr beam splitter. The taranakite sample was mixed with KBr at a weight ratio and was lightly ground in an agate mortar. The mixture was then pressed into a transparent pellet. Infrared spectra were collected in the spectral range of 400–4000 cm−1, averaging 64 scans for both sample and background, with 4 cm−1 nominal resolution. High-temperature data collection was done using a Specac P/N 5850 microfurnace in the range of 298–508 K. The temperature was controlled using a Cr–Al thermocouple placed close to the sample and is accurate within ±1 K according to the manufacturer. Temperature was increased in 5–10 K steps with a heating rate of 10 K min−1, from RT (298 K) to 508 K. At every step, a delay of 30 s was introduced before starting data acquisition to reach temperature equilibrium.
2.5 HT in situ micro-Raman spectroscopy
Raman spectra were measured on selected crystals (∼ µm) of taranakite using a Horiba Explora-Plus single-monochromator spectrometer at the Department of Earth, Environmental and Life Sciences of the University of Genoa. Raman scattering was excited using the 785 nm line of a diode-generated laser and collected via a 50× LWD objective (N.A. 0.5) and a grating of 600 grooves mm−1, leading to a spectral resolution of ∼ 3 cm−1. The instrument was calibrated to the silicon Raman peak at 520.5 cm−1 (T2g mode). Raman spectra under ambient conditions were first collected between 100–4000 cm−1 to check for OH groups or water molecule vibrational contributions, but no signal was detected in our experiments above 2000 cm−1. In order to collect a Raman spectrum of francoanellite at ambient temperature and to use it as a reference for HT in situ Raman spectroscopic analysis, an aliquot of taranakite powder was heated in a static oven at 383 K for 6 h to convert it into francoanellite. After checking that the material had fully transformed via XRPD, the Raman spectrum was collected using the same experimental setup detailed above.
Consequently, HT Raman spectra have been collected between 100 and 2000 cm−1. HT experiments were conducted in air using a Linkam stage TS1500, with a heating rate of 10 K min−1. For non-isothermal runs, the investigated temperature range was between 298–478 K. At every temperature step, a delay of 180 s was introduced before starting data acquisition to avoid thermal gradients in the sample. The acquisition time for each spectrum was 90 s averaged over three accumulations. The isothermal runs were conducted at 353, 358, 363, and 373 K and lasted 143, 155, 59, and 58 min, respectively; the acquisition time was 90 s averaged over three accumulations, for a total of 270 s for each spectrum. The spectra were collected every 5 min (counted between each start of spectral acquisition). Further details on spectral fitting and data evaluation are given in the Supplement (Sect. S1).
3.1 Thermodynamic and experimental determination of the phase transformation temperature
3.1.1 Thermodynamic calculations by the polyhedral approach
The transformation of taranakite into francoanellite can be described according to the following partial dehydration reaction:
At the equilibrium temperature (TEQ), the change in Gibbs free energy (ΔG) associated with the reaction must be equal to zero, i.e.
To calculate the TEQ of the reaction, it is necessary to calculate the contribution of Gibbs free energy of all the phases in the reaction at varying temperatures (and at a constant pressure of 1 bar). La Iglesia (2009) implemented the polyhedral approach in order to calculate the individual contributions to the Gibbs free energy of formation (gi) and to the enthalpy of formation (hi) of ionic groups in a set of 41 phosphate minerals. Following the method of Chermak and Rimstidt (1990), the author provided also the temperature functions (gi,T) of the constituent basic units, as expressed by
which can be simplified to
where
These thermodynamic parameters can be used to calculate the Gibbs free energy of formation (Gf) under the target conditions (temperature T and reference pressure Pr=1 bar) of both taranakite and francoanellite via the summation of the contributions of each polyhedron in the structure (i) multiplied by the stoichiometric coefficient of each unit (n), as stated in Eq. (5):
Therefore, using the thermodynamic dataset of La Iglesia (2009), the Gibbs free energy of taranakite at T and Pr can be obtained as
while that of francoanellite as
Given that all of the water released by the dehydration reactions is in the form of vapour phase, as confirmed by TG-DTA experiments and IR spectroscopic analysis of the evolved gases (Casale et al., 2025), we use the values of Gf of water vapour at P=1 bar as reported by Barin (1995) in the temperature range 298.15–1000 K.
The equilibrium temperature of the phase transformation as obtained by minimising the of Reaction (1) is found to be TEQ=369.12 K (95.97 °C). Experiments conducted at high temperatures making use of XRPD, FTIR, and micro-Raman spectroscopy point to temperature conditions for the onset of the phase transformation which differ by less than 10 K from that predicted by thermodynamic calculations, also providing useful insights into the structural modifications occurring during the transformation, especially regarding the hydrogen-bonding network (see details in the following paragraphs). The good agreement between the calculated and measured values for the phase transformation temperature confirms the ability of the polyhedral approach to correctly predict Gibbs free energies and enthalpies of the formation of phosphates and structurally complex minerals with an uncertainty of less than 1 %–2 % (see Vieillard and Tardy, 1984; La Iglesia, 2009; Kampf et al., 2013, 2018; Kolitsch et al., 2018).
3.1.2 HT XRPD
Selected XRPD scans collected at increasing temperatures in the angular range of 4–45° 2θ are reported in Fig. 2a. The reflections of taranakite do not show significant variations up to 358 K. Above this temperature, their intensities decrease steadily, while the peaks of francoanellite grow quickly in intensity, becoming the only observable peaks at 398 K. This behaviour is also clearly expressed by the shift in the relative intensity of the basal (006) reflections of the two phases, reported in Fig. 2b, centred at 6.47° 2θ (d≈15.85 Å) for taranakite and 7.43° 2θ (d≈13.80 Å) for francoanellite.
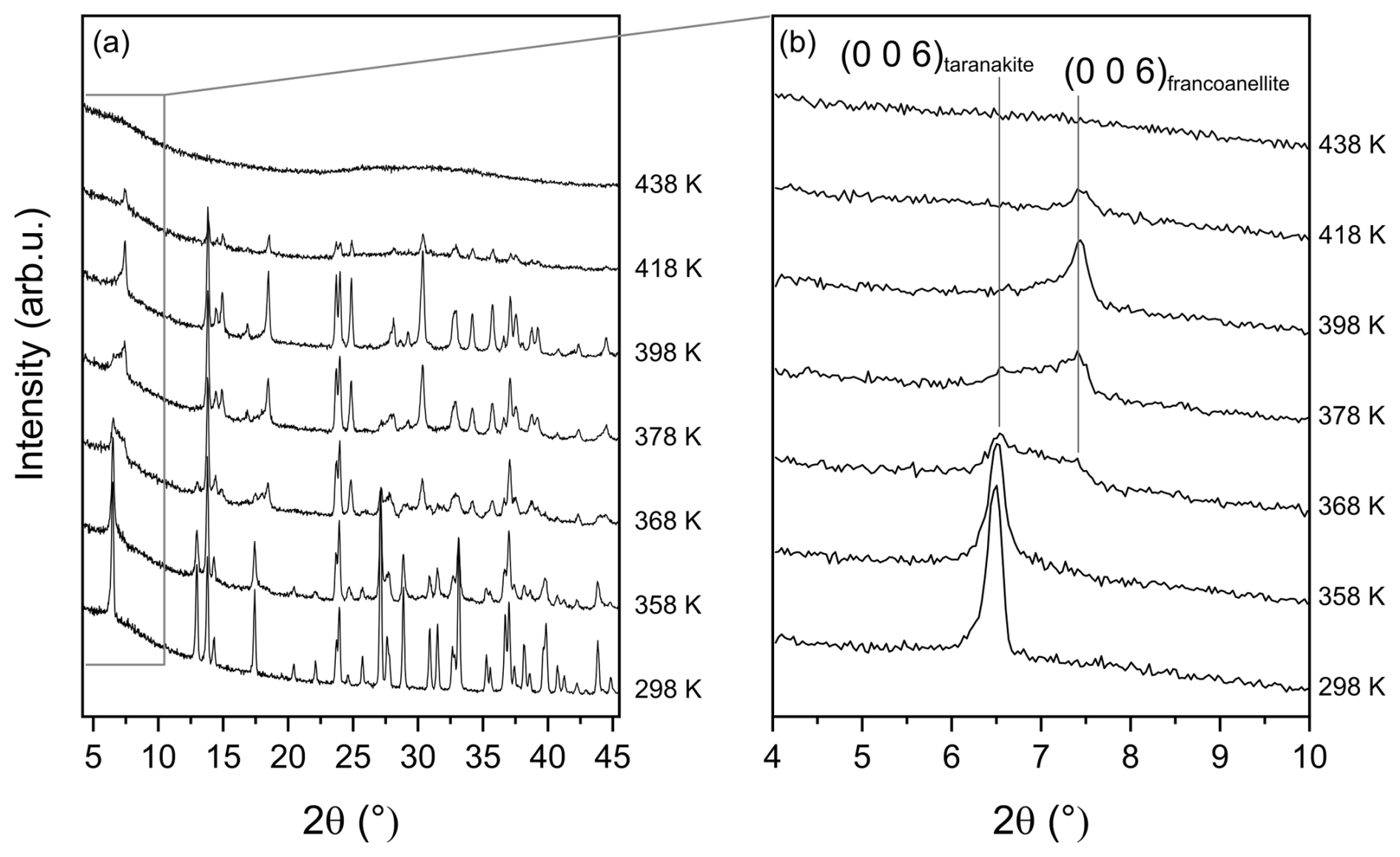
Figure 2Selected HT XRPD scans collected in the range of 298–438 K (a) and detail of the range of 4–10° 2θ characterised by the presence of the basal (006) reflections of the two phases at 6.47° 2θ (d≈15.85 Å) for taranakite and 7.43° 2θ (d≈13.80 Å) for francoanellite (b).
The lattice parameters and cell volume of taranakite exhibit a slight increase due to thermal expansion prior to the onset of dehydration, as reported in Fig. 3a–c, while those of francoanellite, shown in Fig. 3d–f, decrease slowly until reaching a minimum at 408 K before the unit cell rapidly collapses at temperatures higher than 418 K, when the material becomes X-ray amorphous.
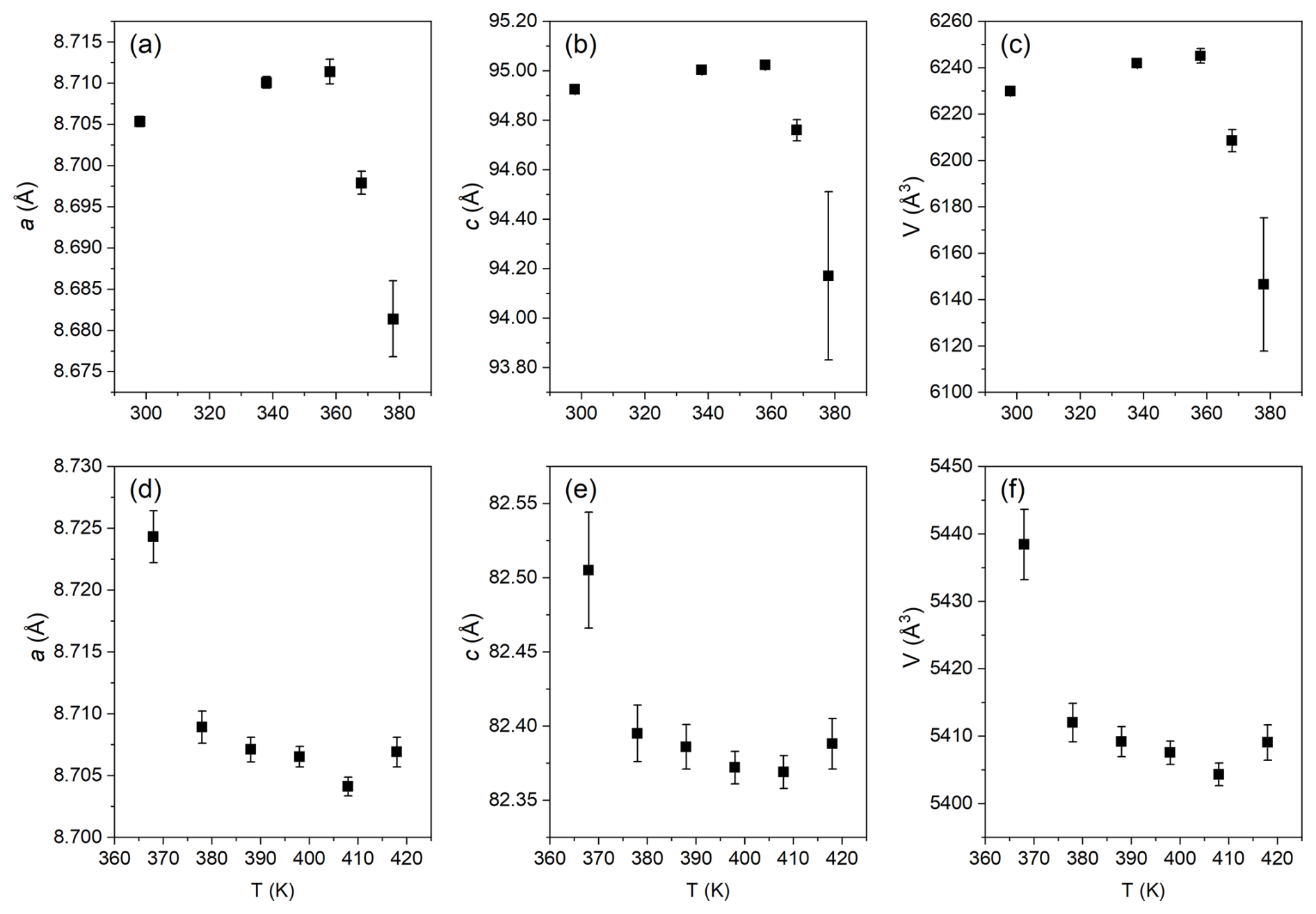
Figure 3Modifications of the lattice parameters of taranakite (a–c) and of francoanellite (d–f) as a function of increasing temperature in the range of 298–418 K, as obtained by Rietveld refinement of the XRPD data.
3.1.3 HT FTIR
The infrared spectrum of taranakite and the spectra collected in situ at increasing temperature via HT FTIR analysis, displayed in Fig. 4a, are quite complex and result from the convolution of the vibrational modes of PO4, PO3OH, NH4, OH groups, and H2O molecules. References from the available literature that support the band assignments, together with a summary of the observed variability of peak positions at different temperatures, are reported in Table 1. As shown in greater detail in Fig. 4b, in the lower wavenumber region (400–670 cm−1), the infrared spectrum of taranakite collected at RT displays six moderately resolved peaks; the first five (415, 437, 454, 550, 602 cm−1) are assigned to the symmetric and antisymmetric bending modes of the phosphate units, while the last one, centred at 643 cm−1, is assigned to the OH librational mode (Ross, 1974). Upon heating, these bands show modest decreases up to 438 K. At higher temperatures, they are rapidly replaced by a very broad band centred at ∼ 480 cm−1. In the region comprised between 670 and 1350 cm−1, the most intense absorption bands of taranakite are observed. The sharp and intense peaks centred at 952, 1019, 1059, 1099, and 1186 cm−1 are assigned to the symmetric and antisymmetric stretching modes of the phosphate and hydrogen phosphate groups, while the weaker bands near to 877 and 904 cm−1 can be attributed to the hydroxyl bending mode (Ross, 1974; Vantelon et al., 2001). In HT experiments, the peaks attributed to OH bending completely disappear at ∼ 400 K, while the phosphate contributions show a slight decrease in intensity. At T>470 K, only two very broad bands centred at 922 and 1126 cm−1 are observed and are attributable to the vibrational modes of PO4 tetrahedra in the amorphous material resulting from the thermal decomposition of francoanellite.
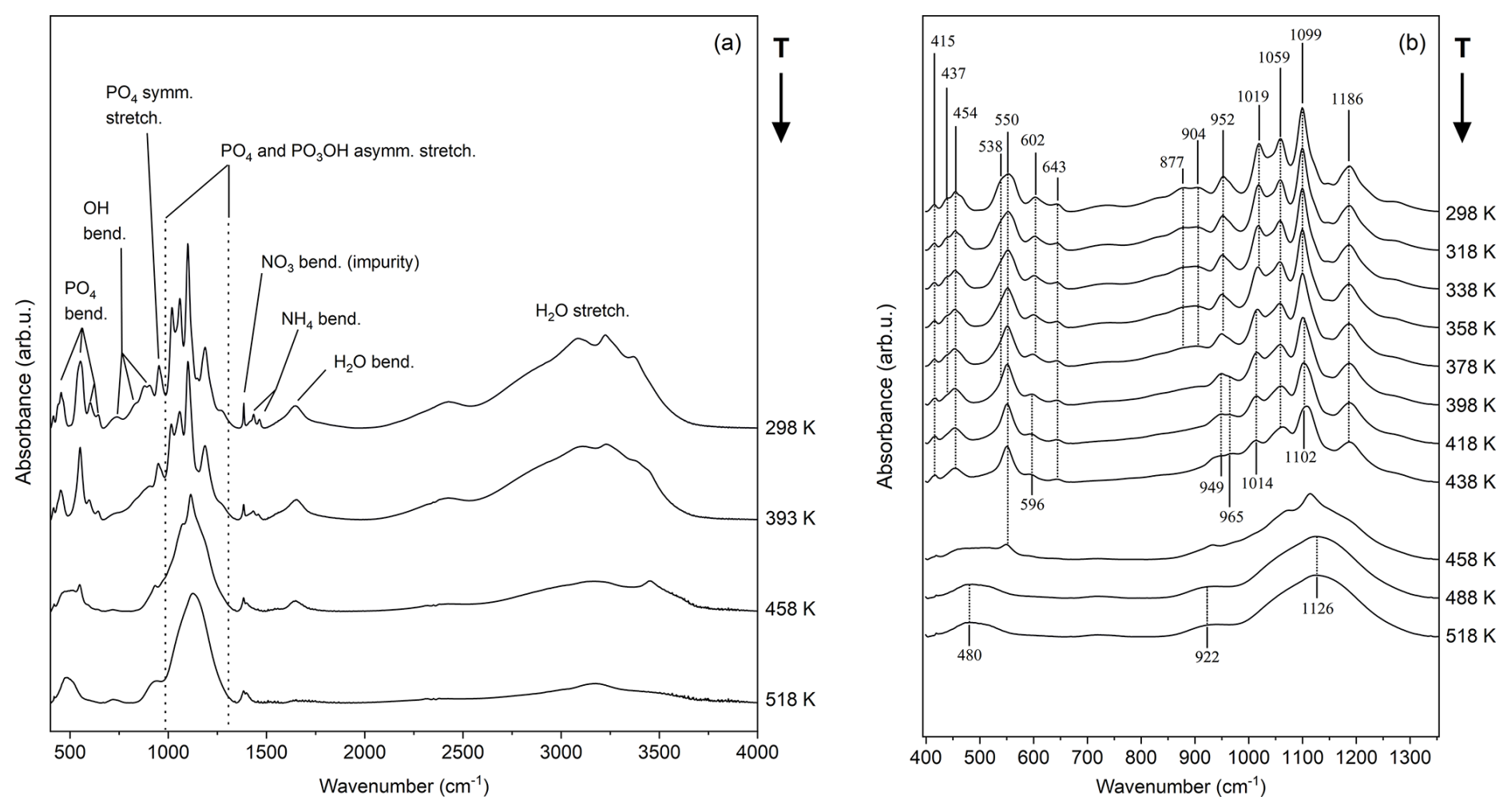
Figure 4Selected HT FTIR spectra collected in situ at increasing temperatures in the range of 400–4000 cm−1 (a) and detail of the 400–1400 cm−1 range, displaying the most important bending and stretching contributions of the PO4 and PO3OH groups (b).
As shown in Fig. 5a, in the range of 1350–2000 cm−1, the absorption bands attributable to the NH4 bending modes (1434 and 1463 cm−1) (Sergeeva et al., 2024) and H2O bending modes (1645 cm−1) are observable. A very sharp peak, centred at 1384 cm−1, is attributed to the presence of nitrate impurities adsorbed on taranakite crystals (Chukanov and Chervonnyi, 2016). At temperatures comprised between 358 and 418 K, all bands show a moderate decrease, while, at higher temperatures, the peaks attributed to NH4 and H2O bending rapidly disappear. The peak attributed to nitrate impurities remains visible with a weak shoulder centred at ∼ 1398 cm−1. The 2000–4000 cm−1 region, shown in Fig. 5b, displays intense and very broad bands, which are attributed to the overlapping contributions of OH and H2O stretching modes. The highest-frequency peak observed in the spectrum of taranakite is centred at 3374 cm−1. Using the correlation of Libowitzky (1999), the bonds with the closest expected frequency of vibration (3390 cm−1, in excess of 16 cm−1) in the structure of taranakite are the HBs connecting the oxygen vertices O(2) shared by both Al(3) octahedra and P(1) tetrahedra to the H of water molecules coordinating Al(2) octahedra, following the sites' denomination as given by Dick and Zeiske (1998). As the temperature increases, the peak near to 3374 cm−1 decreases in intensity and is replaced by a peak centred at about 3450 cm−1, marked by an asterisk in Fig. 5b. The latter is first noticed as a shoulder of the broader contributions at 398 K and remains sharp and visible up to 458 K, in contrast with the other peaks in the same region, which become barely discernable under the broad contribution centred near to 3200 cm−1 at T≥398 K. It is difficult to assign this peak to a specific bond in the structure of francoanellite when basing the choice solely on the correlation of Libowitzky (1999) as the bond with the closest expected frequency of vibration, represented by the same HB considered above for taranakite, should be centred near to 3544 cm−1, in excess of 94 cm−1.
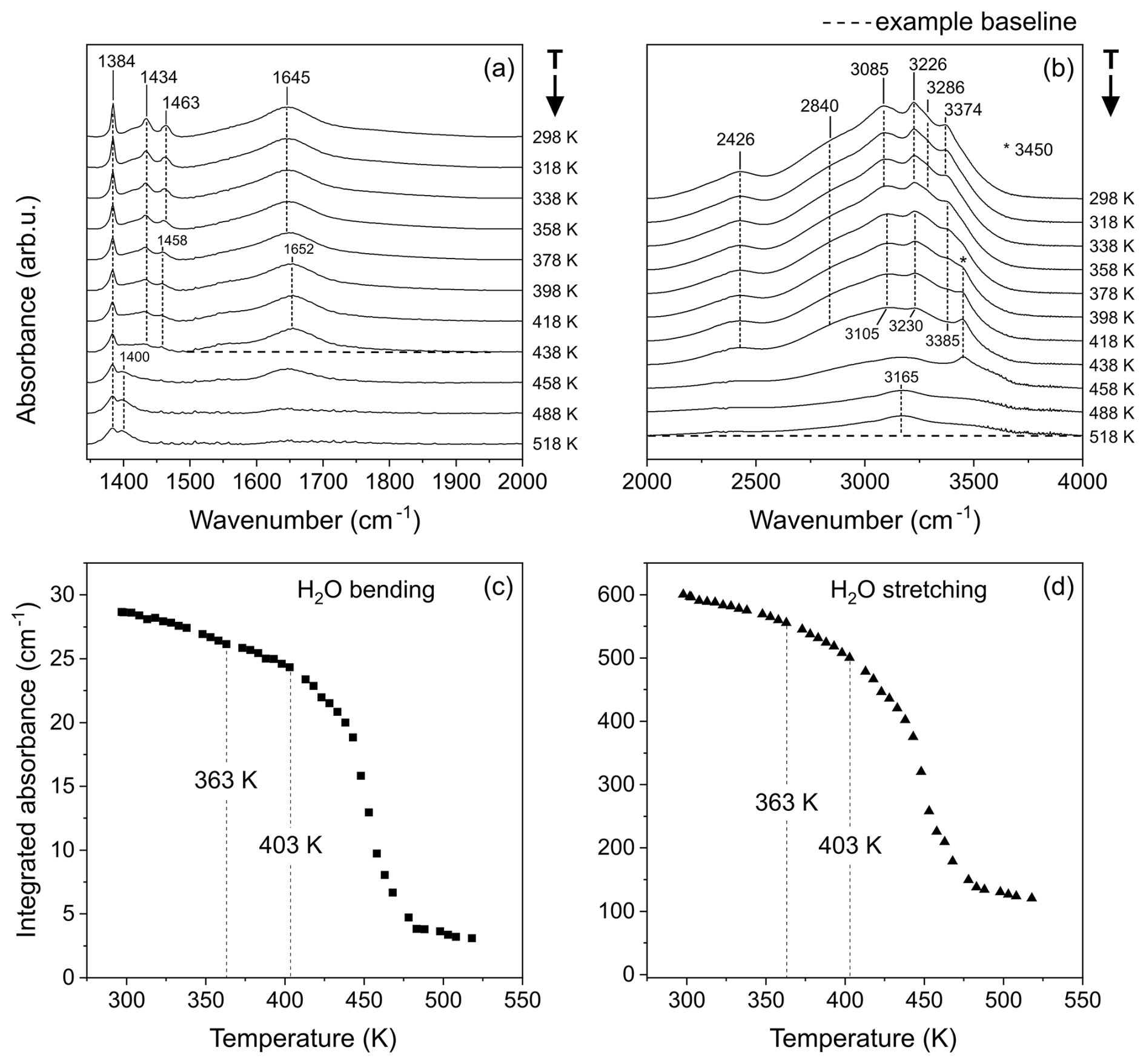
Figure 5Selected FTIR spectra collected at increasing temperatures, represented in the spectral ranges of 1350–2000 cm−1 (a) and 2000–4000 cm−1 (b). Example baselines are shown as dashed black lines connecting the minima that serve as boundaries for integration in the two spectral regions of interest in the ranges of 1495–1952 and 2000–4000 cm−1, respectively. The resulting intensities expressed in terms of integrated area are shown in panel (c) for the H2O bending region and in panel (d) for the H2O stretching region.
The change in absorbance of the H2O bending mode (measured by integrating the area of the region of 1495–1952 cm−1) as a function of increasing temperature is reported in Fig. 5c, while that of the H2O stretching region (measured by integrating the area in the range of 2000–4000 cm−1) is reported in Fig. 5d. The values are obtained from integration of the spectra after correction for a linear baseline connecting the two minima constituting the upper and lower bounds of the selected spectral ranges. The decrease in integrated area is constant up to 363 K, after which a subtle change in slope is visible, which is compatible with the onset of francoanellite formation. The new trend is maintained up to ∼ 403 K, followed by a marked increase in the rate of dehydration, corresponding to the complete expulsion of crystallisation water, collapse of the crystalline structure, and amorphisation of the material, in accordance with the previously outlined HT diffraction data.
Table 1Observed IR fundamental frequencies for taranakite at RT (298 K) and during in situ heating at 398 K and 518 K.
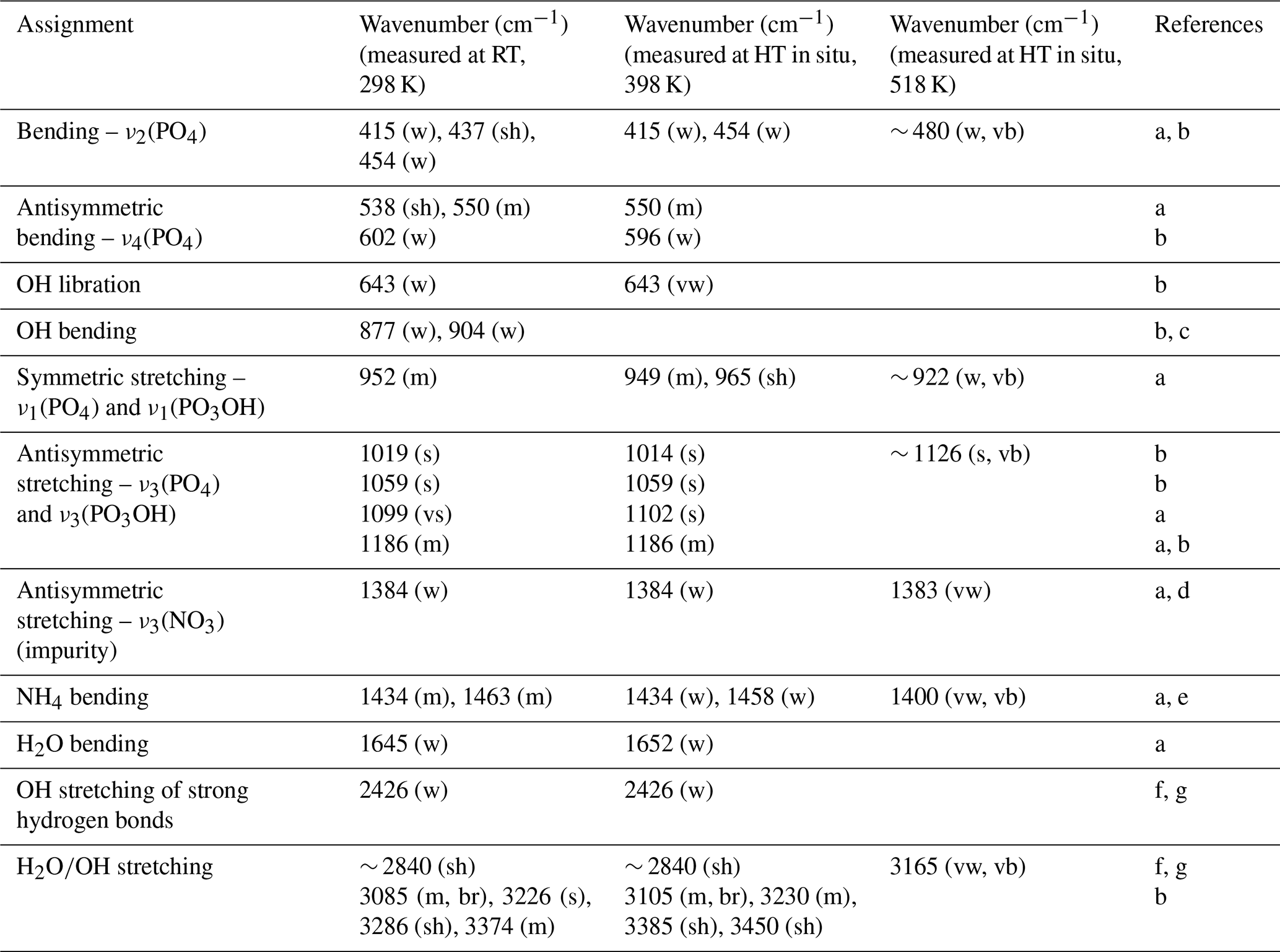
References for peak assignments: a shows Arlidge et al. (1963), b shows Ross (1974), c shows Vantelon et al. (2001), d shows Chukanov and Chervonnyi (2016), e shows Sergeeva et al. (2024), f shows Berry and Baddiel (1967), and g shows Taddei et al. (2000). Abbreviations: vw denotes very weak, w denotes weak, m denotes medium, s denotes strong, vs denotes very strong, sh denotes shoulder, br denotes broad, and vb denotes very broad.
3.1.4 Micro-Raman spectroscopy
The Raman spectrum of taranakite shows relatively strong peaks in the region between 100 and 1600 cm−1, which can be further subdivided into two distinct subregions. The first one, in the range of 100–670 cm−1, shown in Fig. 6a, exhibits peaks which can be attributed to external lattice vibrations and to the bending modes of PO4 and PO3OH groups (Frost et al., 2011). The second one, in the range of 700–1600 cm−1, reported in Fig. 6b, displays strong vibrations attributable to the symmetric and antisymmetric stretching modes of the phosphate and hydrogen phosphate ions, together with very weak and broad peaks; the latter are not assigned to any vibrational mode in this work because of their low intensity and significant uncertainty in peak position. A Raman spectrum of francoanellite was collected at ambient temperature on a sample prepared by static heating of taranakite at 383 K for 6 h. In the range of 100–670 cm−1 (Fig. 6c), the observed spectrum results from a complex convolution of peaks attributable to external lattice modes and to PO4 and PO3OH bending modes – by analogy to the ones of taranakite. In the 700–1600 cm−1 range (Fig. 6d), the Raman spectrum of francoanellite is characterised by the presence of fewer but more intense peaks, assigned to the symmetric and antisymmetric stretching vibrations of PO4 and PO3OH groups, with the most intense peak centred near to 934 cm−1.
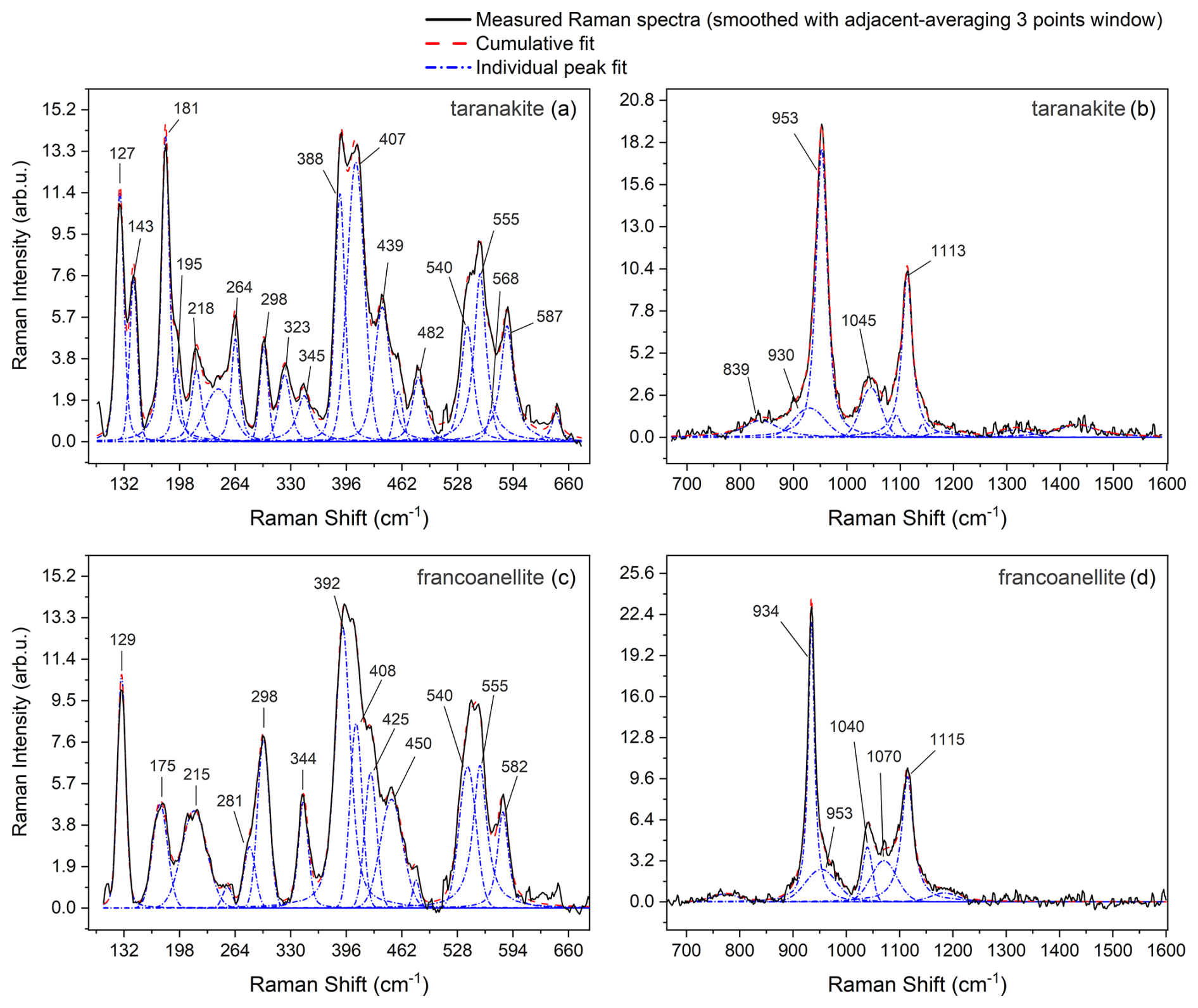
Figure 6Raman spectra collected at RT in the 100–670 cm−1 spectral range (a, taranakite; c, francoanellite) and in the 700–1600 cm−1 range (b, taranakite; d, francoanellite). Labels report the position (approximated to the nearest integer, in units of cm−1) of the main Raman peaks, obtained from the fitting procedure, which was performed using Origin Pro.
All of the observed frequencies obtained via peak fitting for taranakite and francoanellite are summarised in Table 2. The remaining part of the spectrum in the investigated window (up to 4000 cm−1) does not display evidence of Raman scattering.
Raman spectra collected in situ at increasing temperatures allow us to visualise the development of the phase transformation from the point of view of the vibrational contributions of lattice modes and phosphate functional groups. However, it should be mentioned that the borosilicate cover of the HT stage significantly hampers the Raman scattering intensity of already weak peaks that, consequently, were no longer detectable at high temperature. In the range of 100–700 cm−1 (see the Supplement, Fig. S1), at T between 348 and 398 K, the spectra are subject to significant changes in intensity and band position of the lattice modes and phosphate bending modes. At 398 K, the peaks are coincident with those of francoanellite as measured under ambient conditions, although the loss in intensity and overall resolution is noticeable for the reasons mentioned earlier. At T>413 K, all peaks show a marked decrease in intensity and finally disappear at 478 K, pointing to the complete amorphisation of the material.
Table 2Measured wavenumbers for the main peaks of taranakite and francoanellite (spectra collected at 298 K).
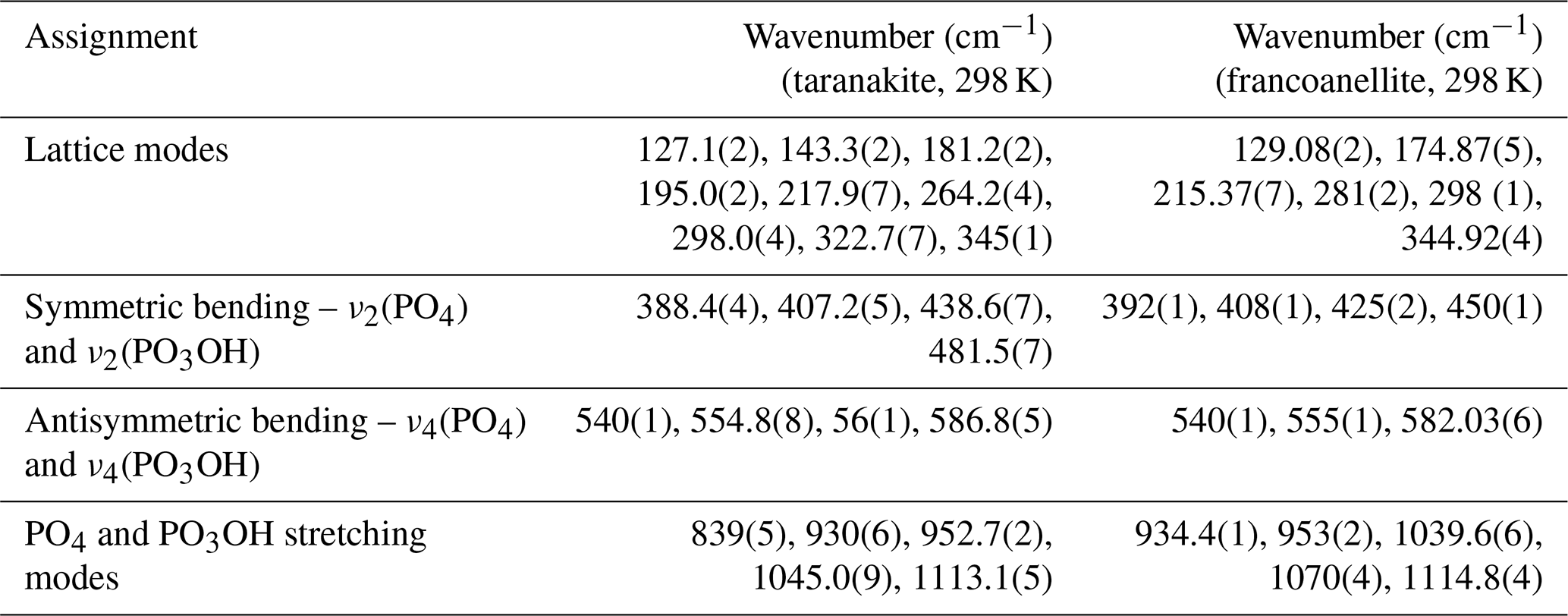
Assignments for taranakite peaks after Frost et al. (2011). The assignments for francoanellite peaks are proposed, by analogy, in relation to those of taranakite.
In the phosphate stretching region (700–1300 cm−1), the most prominent change observed during heating is the decrease in the intensity of the peak near to 952 cm−1, attributed to the symmetric stretching vibrations of phosphate units of taranakite (Frost et al., 2011), coupled with the growth of an intense peak near to 933 cm−1, attributed to the same vibrational mode in the structure of francoanellite (Fig. 7a). According to Popović et al. (2005), the peak position of these phonon modes can be used to precisely estimate the P–O bond length within the different structures. The relation between phonon wavenumber ω and P–O bond length R (±0.01 Å) takes the form of
where cm−1, and pm−2. Using the values of ω of 952 and 933 cm−1, the resulting bond lengths are 1.559 and 1.565 Å, respectively. These values are remarkably comparable to the length of 1.561(5) Å given by Dick and Zeiske (1998) for the P(2)–O(5) bond in isolated P(2) tetrahedra in the layers of the two phases. These orthophosphate tetrahedra are characterised by a complete lack of oxygen sharing with other coordination polyhedra but participate in extensive hydrogen bonding as hydrogen bond acceptors. These observations allow us to confirm that, in this system, the internal vibrational modes of phosphate, most notably the ν1(PO4) peak, are responsive to phase changes and can be used to trace the evolution of the transformation. We therefore followed the evolution of the integrated intensity of the two aforementioned peaks in the T range of 298–393 K. Spectral fitting was performed as exemplified in Fig. 7b; after adequate pre-treatment (as specified in Sect. 2.5), peak fitting was performed in the spectral range of 745–1250 cm−1 using pseudo-Voigt functions to obtain the position, full width at half maximum (FWHM), and integrated intensity of the Raman peaks. The value of the peak shape parameter was allowed to vary in the range between 0 and 1, with the former corresponding to a pure Gaussian peak shape and the latter corresponding to a pure Lorentzian peak shape. Results were discarded whenever the standard errors reached or exceeded half the value of the respective parameter.
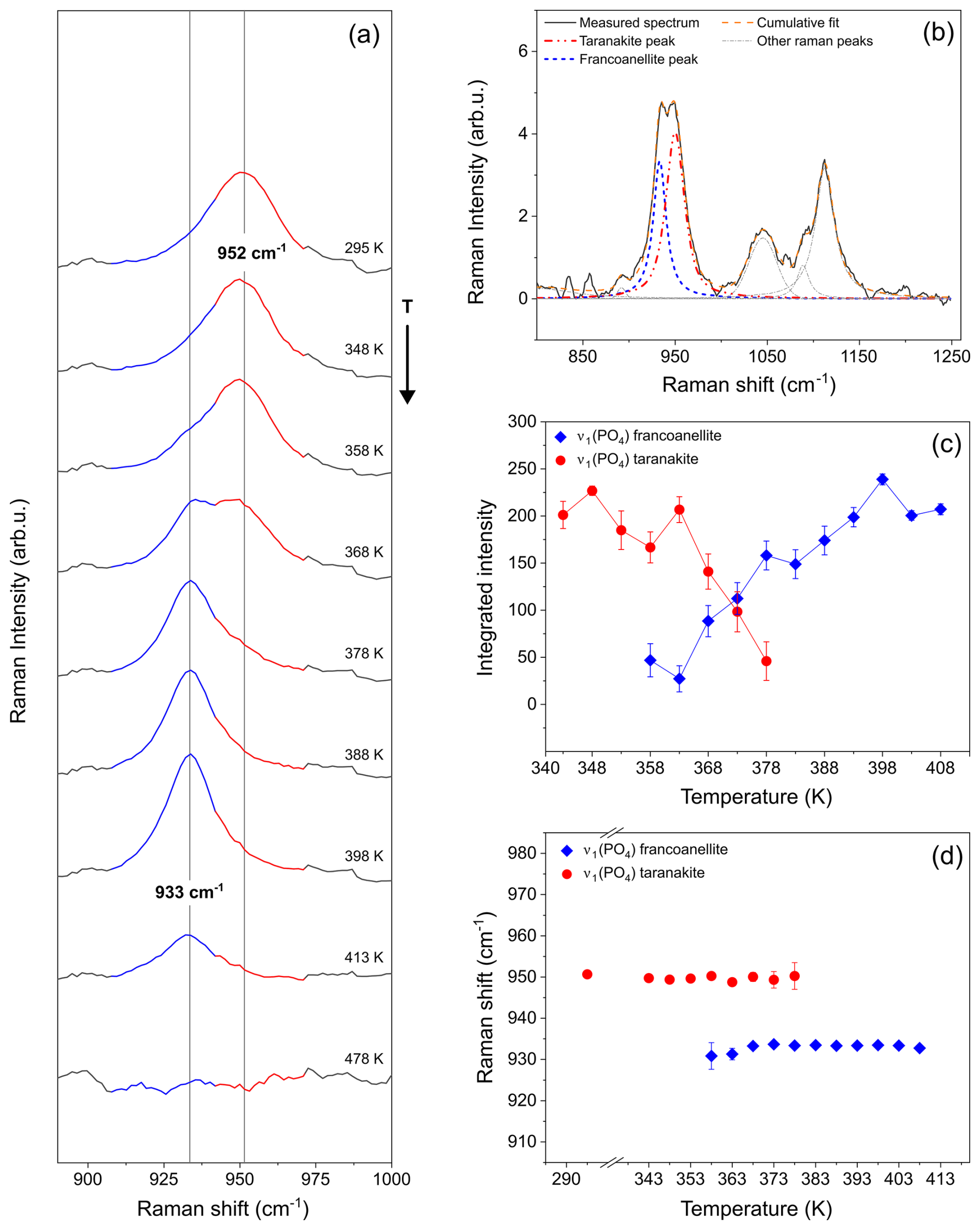
Figure 7Raman spectra collected during in situ heating of a taranakite microcrystal (a), showing the changes in the relative intensities of the phosphate stretching peaks near to 952 and 933 cm−1, assigned to, respectively, taranakite and francoanellite. The vertical lines and the coloured regions of the spectra in this panel are a guide to the eye. (b) Example of peak fitting, which allowed us to trace the evolution in intensity (c) and peak position (d) of the two peaks. Data used to produce these graphs are available in the Supplement (Tables S1–S2).
The resulting peak positions and integrated intensities are reported in Fig. 7c–d. Upon heating, the taranakite peak near to 952 cm−1 displays a subtle shift to a lower frequency and a significant decrease in intensity for T>358 K. At the same T, a sharp increase in the integrated intensity of the francoanellite peak near to 933 cm−1 is observed. No significant change in peak position is observed within the investigated T range for both peaks. At T>378 K, only the francoanellite peak is observed, while the taranakite peak is no longer present.
3.2 Determination of the empirical activation energy (Ea) from time-resolved isothermal HT micro-Raman experiments
HT micro-Raman experiments conducted under isothermal conditions at 353, 358, 363, and 373 K allowed us to follow the evolution of the phase transformation with time. In order to visualise the rate of the taranakite-to-francoanellite transformation process, the evolution of the integrated intensity values of the distinctive peaks of the two phases at ∼ 952 cm−1 and 933 cm−1, attributed to the ν1(PO4) symmetric stretching modes of P(2)–O(5) bonds, was monitored. The intensity values were obtained via peak fitting of the spectra acquired at fixed temperatures and varying times. For each time-resolved data set, the first acquired spectrum (at t=3 min) was fit without constraining the wavenumbers of the peaks; in the following spectra (from t=8 min to the end), the wavenumber of the taranakite ν1(PO4) was fixed at the value resulting from the fit of the first spectrum. The variations in the integrated intensities as a function of time are shown in Fig. 8. The complete set of data used to produce the figures in this section is reported in the Supplement (Tables S3–S10).
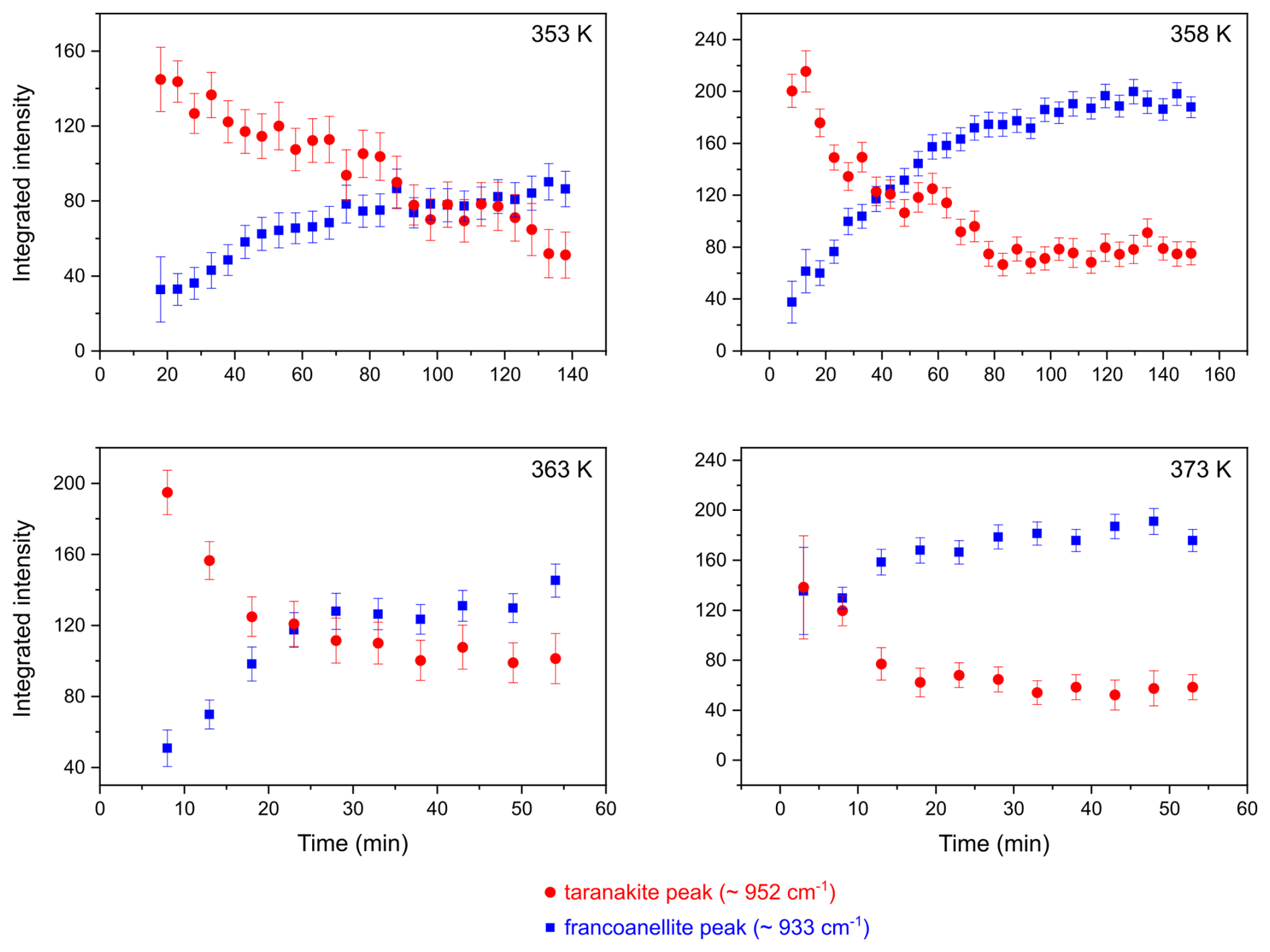
Figure 8Variations in integrated intensity as a function of time for the PO4 symmetric stretching peaks of taranakite and francoanellite, as obtained from peak fitting of spectra collected at HT in isothermal conditions.
The reaction product fraction α has been calculated from the integrated intensity of the two Raman peaks as follows:
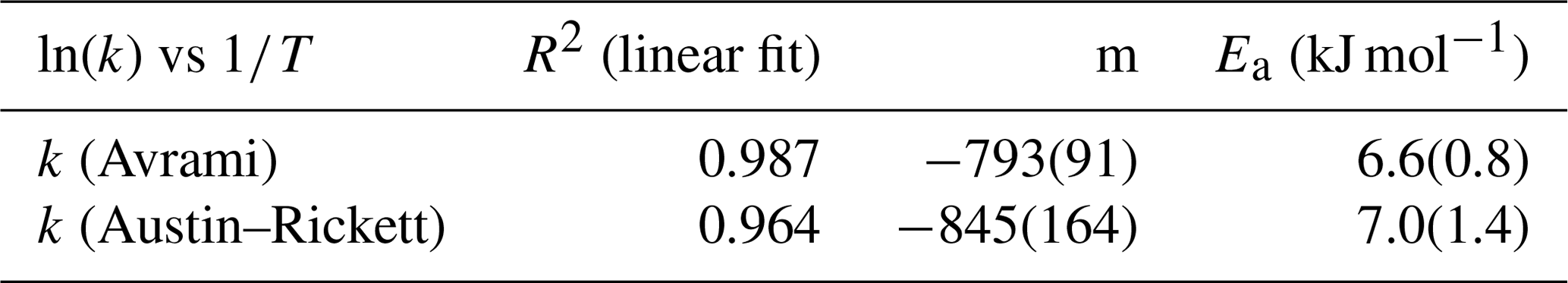
where Itrk and Ifrn are the integrated intensity values of the taranakite and francoanellite ν1(PO4) peaks, respectively. The fraction transformed as a function of time is shown via an Avrami plot in Fig. 9a and an Austin–Rickett plot in Fig. 9b. The linear fit of the experimental data collected between 353 and 363 K shows slope coefficients that are constant within uncertainty (see Table 3). On the other hand, the linear fit of the data measured at 373 K shows a slope coefficient that is significantly different with respect to those obtained at lower T. Furthermore, the isothermal data collected between 353 and 358 K show deviations from linearity when approaching the highest values of the fraction transformed, suggesting that the kinetic mechanism regulating the reaction is also dependent on time and on the relative abundance of reactants and products (Redfern, 1987). The reaction rates (k) as calculated from the slope (n) and the intercept (nln k) of the linear fits of the experimental data collected at different temperatures are given in Table 3 and are shown in the Arrhenius plot in Fig. 9c as a function of . To calculate the activation energy Ea of the taranakite-to-francoanellite transformation, we have selected the data within the temperature range in which the reaction appears to be isokinetic (i.e. between 353 and 363 K). The results are summarised in Table 4.
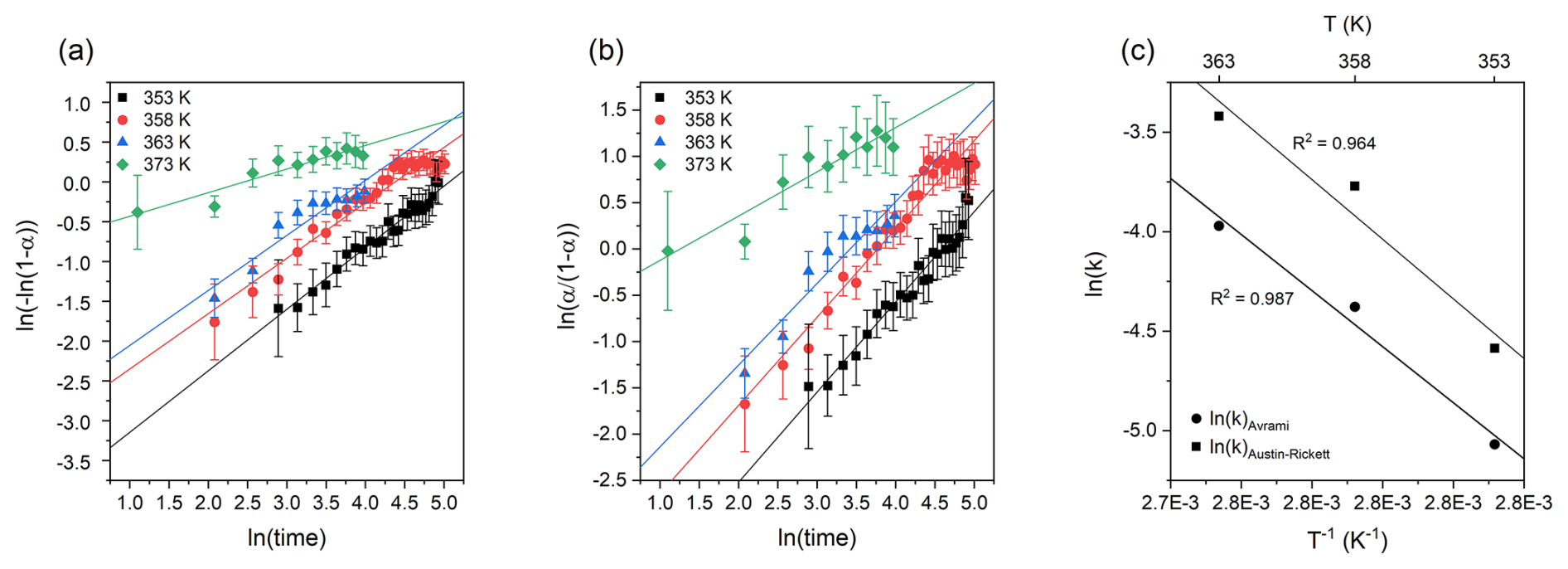
Figure 9Avrami plot of the fraction transformed (a) and Austin–Rickett plot of the fraction transformed (b); vertical segments represent the calculated uncertainty α. Arrhenius plot ln (k) vs. T−1 (c) and linear fits used to estimate the empirical activation energy values.
Table 3Reaction rates calculated using the Avrami and Austin–Rickett methods.
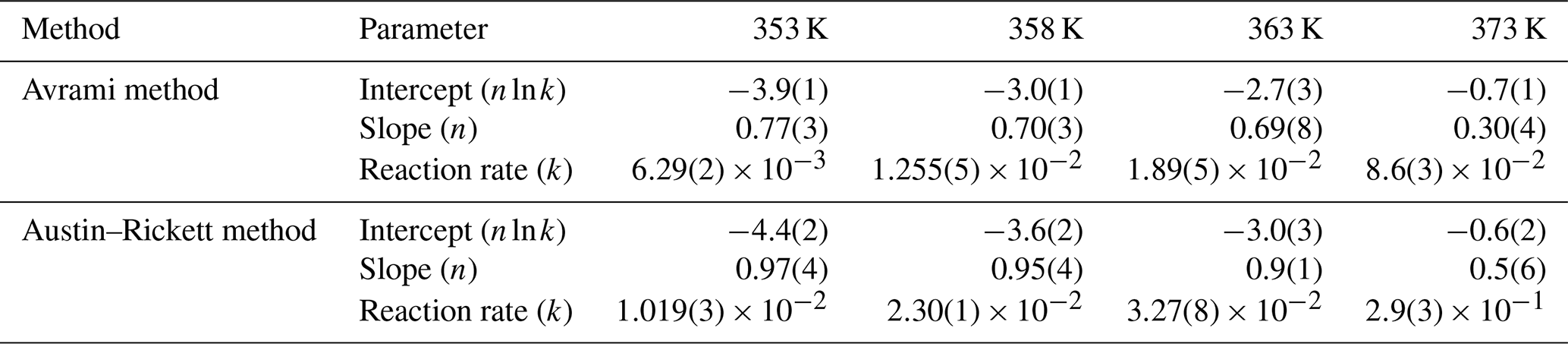
Table 4Values of slope (m) and empirical activation energy (Ea) as derived from the application of the Avrami and Austin–Rickett methods.
To check if the deviations from linearity visible in both the Avrami and Austin–Rickett plots may significantly affect the calculated Ea, we performed the calculation with the “time to a given fraction” approach, which has the advantage of being independent of rate functions (Bray and Redfern, 1999, and references therein) (Fig. 10). For a fraction transformed of 0.3 in the temperature range of 353–363 K, the resulting Ea is 7.6 ± 0.7 kJ mol−1, in good agreement with the results obtained from the application of the Avrami and Austin–Rickett methods. A further attempt for a fraction of 0.55 leads to an Ea of 7.2 ± 1.9 kJ mol−1, proving the consistency of the approach.
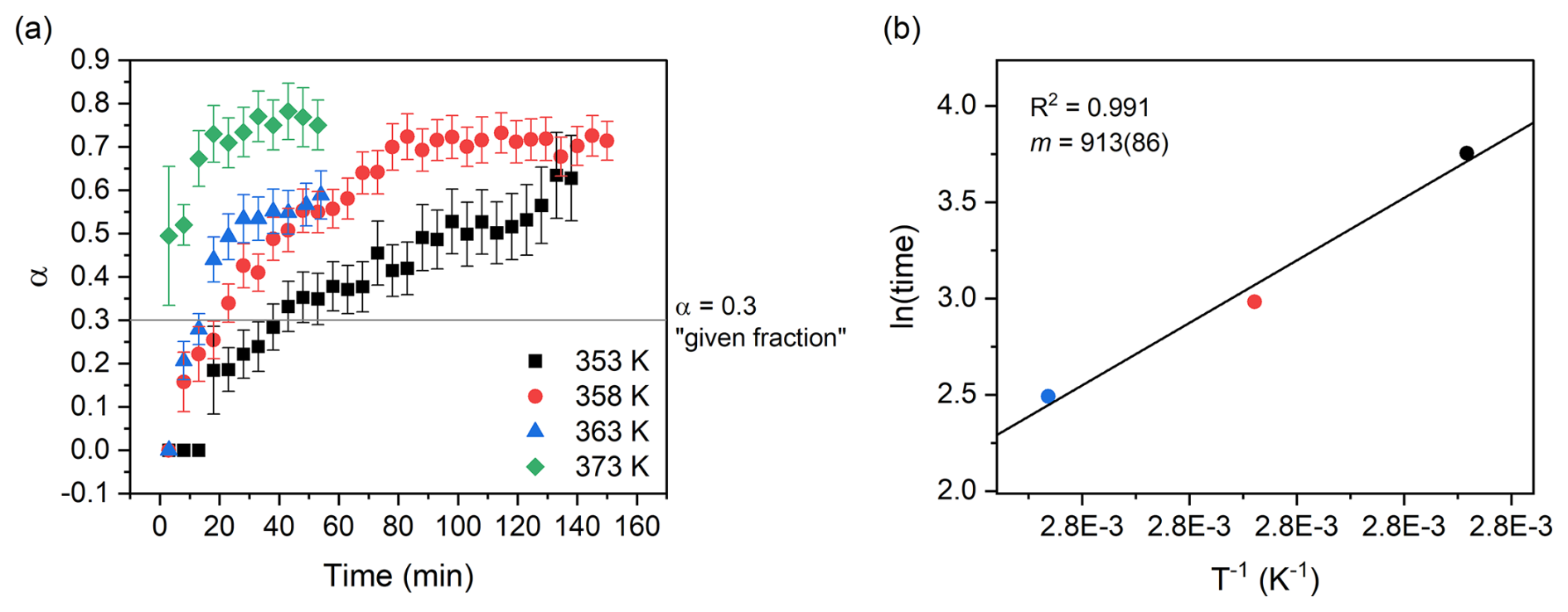
Figure 10Fraction transformed versus time plot (a); the horizontal line indicates the 0.3 fraction used for the “time to a given fraction” method, while vertical segments represent the calculated uncertainty for α. The times deriving from the isothermal runs of the isokinetic range (353–363 K) were plotted on a ln (time) vs. plot (b), and the slope of the linear best fit was used to calculate the empirical activation energy of the transformation.
3.3 Considerations regarding the occurrence of francoanellite in cave environments
Justifying the occurrence of partially dehydrated phosphates and sulfates (i.e. bassanite, monetite, francoanellite) in the parageneses deriving from guano degradation in caves remains a complex task. Although it is often postulated that the decomposition of bat guano generates heat, few direct measurements of T at varying depths in guano deposits are present in the available literature. As an example, the in situ temperature measurements in an actively accumulating and decomposing guano mound in Mulu Cave (Borneo) down to a depth of 70 cm, performed by McFarlane and Lundberg (2024), seldom exceeded 30 °C (308.15 K). It is therefore highly unlikely that the dehydration reaction leading to francoanellite formation is regulated by an equilibrium process as the calculated temperature of equilibrium is close to 369 K (95.85 °C) and must be governed by kinetic factors.
The activation energy needed for the onset of the taranakite-to-francoanellite transformation is very low (ca. 7.6 kJ mol−1) if compared to that of other hydrated aluminophosphates such as variscite (Arjona and Franco, 1973), minerals with a layered structure and water occupying interlayer sites such as montmorillonites (Bray and Redfern, 1999; Zabat and Van Damme, 2000), and minerals with loosely bound water molecules held in interstices via H bonds such as zeolites (Majchrzak-Kucęba and Nowak, 2004), which all have activation energies associated with the dehydration reaction >30 kJ mol−1. This finding could justify the partial dehydration of taranakite taking place in cave environments where the heat produced by organic matter degradation causes a slight raise in temperature that is protracted over relatively long periods of time, i.e. tens to hundreds of years under warm and moist conditions (Lundberg and McFarlane, 2024; McFarlane and Lundberg, 2024). Other environmental or anthropogenic factors, such as the presence of powerful halogen spotlights in touristic caves (e.g. in the caves of Castellana), could have an impact on the triggering of dehydration reactions. The low values of activation energy resulting from our experiments are also coherent with the fact that the water molecules involved in the studied transformation are not part of cation hydration shells. Rather, they form an interlayer in the structure of taranakite and are lightly bound to phosphate tetrahedra that reside at the margins of the layers by means of HBs with mean O⋯O distances >2.70 Å and angles >170°, making them classifiable as weak HBs (Libowitzky, 1999), thus representing the weakest link in the chain in terms of structural stability of the phase. As soon as these bonds are broken due to thermal instability, the remaining HBs that link water molecules in the interlayer to water molecules coordinating Al octahedra in the layers are not enough to retain the interlayer–layer cohesion. This type of transformation is reconstructive, but the only bonds which need to be re-established are the HBs holding the layers together in the structure of francoanellite. The obvious topotaxy of the reaction and the complete conservation of the structure layers contribute to keeping the energy barrier low. From the values of the slope (Table 3) of the trends in the Avrami and Austin–Rickett plots (all lower than 1), it is possible to infer that the kinetic mechanism regulating the dehydration is diffusive, with water molecules leaving the crystals by moving parallel to (001) along the interlayer.
Although this demonstrates the feasibility of the transformation of taranakite into francoanellite via partial dehydration in natural environments, other mechanisms should be considered. The most straightforward would be the direct precipitation of francoanellite from percolating solutions. Another possible pathway leading to francoanellite precipitation could stem from RH increase and condensation phenomena causing the dissolution of taranakite, followed by RH decrease and high evaporation rates promoting the crystallisation of francoanellite, as observed in other systems such as the Na–S–O–H one, where, at sufficiently low RH and room temperature, metastable thenardite (NaSO4) precipitates instead of mirabilite (NaSO4 ⋅ 10H2O) (Rodriguez-Navarro et al., 2000).
It must be noted that taranakite transforms into francoanellite within a timescale of only a few tens of minutes if heated to temperatures as low as 358 K. Since those conditions are commonly achieved during routine XRPD sample preparation procedures (Soukup et al., 2008), special care must be taken when preparing phosphate-bearing samples. Heating to remove environmental moisture should also be avoided to prevent the alteration of hydrated phases.
For this work, we conducted an investigation into the thermodynamics and kinetics that define the taranakite-to-francoanellite phase transformation. The equilibrium temperature of the of the reaction, which features a single step of dehydration coinciding with the removal of water interlayers from the structure, was calculated at 369 K. The application of temperature resolved in situ XRPD, FTIR spectroscopy, and micro-Raman spectroscopy gave experimental confirmation of this temperature in a range of ±10 K. The use of HT micro-Raman and HT FTIR spectroscopy allowed us to gather complementary information about the dehydration process of taranakite. While FTIR allowed us to measure T-induced water loss occurring first in taranakite and then in francoanellite before reaching the complete amorphisation of the material, micro-Raman measurements conducted under conditions of increasing T were found to be very sensitive to the taranakite-to-francoanellite transformation, monitored via the changes in the Raman scattering intensity arising from the ν1PO4 stretching modes of the two phases (near to 952 cm−1 in taranakite and 933 cm−1 in francoanellite). Thanks to in situ isothermal micro-Raman analysis, the kinetics involved in the reaction and the associated activation energy were confirmed to be dependent on temperature and, to a lesser extent, on the fraction of reactant transformed. In the inspected temperature range, a field in which the transformation appears to be isokinetic was found and allowed for the estimation of an activation energy (Ea) of 7.6 ± 0.7 kJ mol−1 by means of the “time to a given fraction” kinetic model-free approach, while the values calculated from the application of the Avrami and Austin–Rickett methods were found to be comparable if the range of uncertainty is considered.
The aim of this study was to expand the current knowledge regarding the mineral transformation processes taking place in secondary phosphate deposits deriving from bat guano degradation in cave environments. Contributions shedding light on the stability of authigenic phosphates and on the environmental or intrinsic controls that regulate their transformation are of the utmost importance in improving our understanding of the minerogenetic processes involved in their formation. We hope that the renewed interest in biogenic corrosion and mineralisation processes connected to the presence of bats in caves will foster further research on this topic.
All experimental data are available upon request to the first author.
The supplement related to this article is available online at https://doi.org/10.5194/ejm-38-353-2026-supplement.
CC, FB, JDW, and YG conceptualised the work. YG, NC, FB, and CC designed the experiments and carried them out. DB carried out the thermodynamical calculations. All of the authors participated in the validation process. YG was responsible for visualisation and wrote the original draft. CC and FB supervised the research. All of the co-authors provided critical review and editing of the original paper in the pre-publication stages.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
The authors wish to thank the referees, Bogdan P. Onac and Daniel Atencio, for providing constructive reviews of the paper; the editor-in-chief, Carlos Rodriguez-Navarro, and the associate editor, Encarnación Ruiz Agudo, are acknowledged for the professional handling of the paper during the peer review process and for providing valuable insights which allowed us to broaden the discussion.
Marco Favero and Gabriella Salviulo are acknowledged for granting access to the X-ray diffraction laboratory of the Department of Geosciences of the University of Padua and for conducting the HT XRPD analysis. Members of Gruppo Speleologico CAI Bolzaneto are acknowledged for their support during the sampling (November 2022). Laura Negretti and Roberto Badano are acknowledged for providing support in the sample preparation during the preliminary characterisation of the samples. Tudor Tamaş is acknowledged for engaging in valuable discussions with the first author during his stay at University Babeş-Bolyai (Cluj-Napoca, Romania) during the Spring of 2025.
This research has been partially supported by the University of Genoa (grant agreement no. 100022-2024-FRA-2024).
This paper was edited by Encarnacion Ruiz-Agudo and reviewed by Bogdan P. Onac and Daniel Atencio.
Arjona, A. M. and Franco, M. A.: Kinetics of the thermal dehydration of variscite and specific surface area of the solid decomposition products, J. Therm. Anal., 5, 319–328, 1973.
Arlidge, E. Z., Farmer, V. C., Mitchell, B. D., and Mitchell, W. A.: Infra-red, X-ray and thermal analysis of some aluminium and ferric phosphates, J. Appl. Chem., 13, 17–27, 1963.
Audra, P., De Waele, J. H. A., Bentaleb, I., Chroňáková, A., Krištůfek, V., D'Angeli, I. M., Carbone, C., Madonia, G., Vattano, M., Scopelliti, G., Cailhol, D., Vanara, N., Temovski, M., Bigot, J.Y., Nobecourt, J.C., Galli, E., Rull, F., and Sanz-Arranz, A.: Guano-related phosphate-rich minerals in European caves, Int. J. Speleol., 48, 75–105, 2019.
Audra, P., Heresanu, V., Barriquand, L., Boutchich, M. E. K., Jaillet, S., Pons-Branchu, E., Bosak, P., Cheng, H., Edwards, R.L., and Renda, M.: Bat guano minerals and mineralization processes in Chameau Cave, Eastern Morocco, Int. J. Speleol., 50, 91–109, 2021.
Audra, P., De Waele, J., Uggeri, A., Cailhol, D., D’Angeli, I. M., Fiorucci, A., Foianini, I., Foianini, S., Braucher, R., Inglese, M., Maconi, A., Spreafico, F., Barile, M., Carbone, C., and Tognini, P.: High alpine preglacial caves modified by glacial processes and late condensation-corrosion in the Scerscen Valley (Valmalenco, Western Alps, Italy), Geomorphology, 492, 110054, https://doi.org/10.1016/j.geomorph.2025.110054, 2026.
Badino, G., Calaforra, J. M., Forti, P., Garofalo, P., and Sanna, L.: The present day genesis and evolution of cave minerals inside the Ojo de la Reina Cave (Naica Mine, Mexico), Int. J. Speleol., 40, 125–131, 2011.
Balenzano, F., Dell'Anna, L., and Di Pierro, M.: Ricerche mineralogiche su alcuni fosfati rinvenuti nelle Grotte di Castellana (Bari): strengite alluminifera, vivianite, taranakite, brushite e idrossiapatite, R. C. Soc. Ital. Mineral. Petrol., 30, 543–573, 1974.
Balenzano, F., Dell'Anna, L., and Di Pierro, M.: Francoanellite, H6K3Al5(PO4)8 ⋅ 13H2O, a new mineral from the caves of Castellana, Puglia, Southern Italy, N. Jb. Miner. Mh., 2, 49–57, 1976.
Balenzano, F., Dell'Anna, L., and Di Pierro, M.: Francoanellite from the “Grotta della Rondinella” (Swallow cave) in Apulia (southern Italy): a new occurrence and new data, N. Jb. Miner. Mh., 8, 363–372, 1979.
Barin, I.: Thermochemical Data of Pure Substances, third edn., Wiley-VCH, Weinheim, Germany, 1936 pp., https://doi.org/10.1002/9783527619825, 1995.
Barriquand, L., Bigot, J. Y., Audra, P., Cailhol, D., Gauchon, C., Heresanu, V., Jaillet, S., and Vanara, N.: Caves and bats: morphological impacts and archaeological implications. The Azé prehistoric cave (Saône-et-Loire, France), Geomorphology, 388, 107785, https://doi.org/10.1016/j.geomorph.2021.107785, 2021.
Bernardini, S., Bellatreccia, F., Columbu, A., Vaccarelli, I., Pellegrini, M., Jurado, V., Del Gallo, M., Saiz-Jimenez, C., Sodo, A., Millo, C., Jovane, L., and De Waele, J.: Morpho-Mineralogical and Bio-Geochemical Description of Cave Manganese Stromatolite-Like Patinas (Grotta del Cervo, Central Italy) and Hints on Their Paleohydrological-Driven Genesis, Frontiers in Earth Science, 9, 642667, https://doi.org/10.3389/feart.2021.642667, 2021.
Berry, E. E. and Baddiel, C. B.: Some assignments in the infra-red spectrum of octacalcium phosphate, Spectrochim. Acta A.-M., 23, 1781–1792, 1967.
Bray, H. J. and Redfern, S. A. T.: Kinetics of dehydration of Ca-montmorillonite, Phys. Chem. Miner., 26, 591–600, 1999.
Cancian, G.: I fosfati della “Grotta ad Est di San Martino del Carso” (Gorizia): brushite, idrossiapatite, francoanellite, taranakite, Atti III Covegno Triveneto di Speleologia, 17–18 November 1984, Vicenza, Italy, 1–8, https://www.boegan.it/wp-content/uploads/2023/12/CANCIAN-Graziano-1984-I-fosfati-della-Grotta-ad-est-di-San-Martino-del-Carso-Gorizia.pdf (last access 20 February 2026), 1984.
Casale, M., Galliano, Y., Carbone, C., and Cardinale, A. M.: Ammonia Content in Natural Taranakite: An Experimental Study of Thermal Stability, Crystals, 15, 378, https://doi.org/10.3390/cryst15040378, 2025.
Chermak, J. A. and Rimstidt, J. D.: Estimating the thermodynamic properties (ΔG°f and ΔH°f) of silicate minerals at 298 K from the sum of polyhedral contributions, Am. Mineral., 74, 1023–1031, 1989.
Chermak, J. A. and Rimstidt, J. D.: Estimating the free energy of formation of silicate minerals at high temperatures from the sum of polyhedral contributions, Am. Mineral., 75, 1376–1380, 1990.
Chiorboli, S.: Dati mineralogici preliminari sulla francoanellite della “Grotta Due Piani”, San Martino del Carso (Gorizia), Ist. Miner. Università di Ferrara, Ferrara, Italy, 1984.
Chukanov, N. V. and Chervonnyi, A.D.: Infrared Spectroscopy of Minerals and Related Compounds, Springer, 1109 pp., https://doi.org/10.1007/978-3-319-25349-7, 2016.
Dandurand, G., Duranthon, F., Jarry, M., Stratford, D. J., and Bruxelles, L.: Biogenic corrosion caused by bats in Drotsky's Cave (the Gcwihaba Hills, NW Botswana), Geomorphology, 327, 284–296, 2019.
Dell'Anna, L., Fiore, S., and Laviano, R.: I fosfati di cavità carsiche pugliesi: giacitura, natura e genesi, in: Atti XV Congresso Nazionale di Speleologia, XV Congresso Nazionale di Speleologia, Castellana Grotte, Bari, Italy, 10–13 September 1987, 171–187, 1989.
Dick, S. and Zeiske, T.: Francoanellit K3Al5(HPO4)6(PO4)2 ⋅ 12H2O: Struktur und Synthese durch topochemische Entwässerung von TaranakitFrancoanellite K3Al5(HPO4)6(PO4)2 ⋅ 12H2O: Structure and Synthesis by Topochemical Dehydration of Taranakite, Z. Naturforsch. B, 53, 711–719, 1998.
Dick, S., Goßner, U., Weiß, A., Robl, C., Großmann, G., Ohms, G., and Zeiske, T.: Taranakite – the mineral with the longest crystallographic axis, Inorg. Chim. Acta, 269, 47–57, 1998.
Döbelin, N. and Kleeberg, R.: Profex: a graphical user interface for the Rietveld refinement program BGMN, Applied Crystallogr., 48, 1573–1580, 2015.
Dumitraş, D.-G. and Marincea, Ş.: Sequential dehydration of the phosphate–sulfate association from Gura Dobrogei Cave, Dobrogea, Romania, Eur. J. Mineral., 33, 329–340, https://doi.org/10.5194/ejm-33-329-2021, 2021.
Elliott, P., Kampf, A. R., and Wade, B. P.: Macivorite, IMA 2024-083, CNMNC Newsletter 84, Mineral. Mag., 89, https://10.1180/mgm.2025.22, 2025.
Farrant, A. R., Koether, J. M., Barton, H. A., Lauritzen, S. E., Pennos, C., Smith, A. C., White, J., McLeod, A., and Eavis, A. J.: Pervasive speleogenetic modification of cave passages by nitrification of biogenic ammonia, Geomorphology, 109822, https://doi.org/10.1016/j.geomorph.2025.109822, 2025.
Filippelli, G. M.: Phosphate rock formation and marine phosphorus geochemistry: the deep time perspective, Chemosphere, 84, 759–766, 2011.
Forti, P.: Biogenic speleothems: an overview, Int. J. Speleol., 30, 4, https://doi.org/10.5038/1827-806X.30.1.4, 2001.
Frost, R. L., Xi, Y., Palmer, S. J., and Pogson, R. E.: Vibrational spectroscopic analysis of taranakite (K,NH4)Al3(PO4)3(OH) ⋅ 9(H2O) from the Jenolan Caves, Australia, Spectrochim. Acta A, 83, 106–111, 2011.
Hawthorne, F. C.: Structure and chemistry of phosphate minerals, Mineral. Mag., 62, 141–164, 1998.
Hazen, R. M.: Comparative crystal chemistry and the polyhedral approach, Rev. Mineral., 14, 317–345, 1985.
Hazen, R. M.: A useful fiction: polyhedral modeling of mineral properties, Am. J. Sci., 288, 242–269, 1988.
Hazen, R. M. and Morrison, S. M.: On the paragenetic modes of minerals: A mineral evolution perspective, Am. Mineral., 107, 1262–1287, 2022.
Hector, J. and Skey, W.: Supplementary report on Class 1: coals, building, stones, basaltic rocks, limestones, freestones, metals and metallic ores, magnesian rocks, graphite, clays, cements, economic minerals, bitumen, resins. Appendix A in: New Zealand Exhibition, 1865, Reports and Awards of the Jurors and Appendix, Dunedin, New Zealand, 371–452, https://books.google.it/books?id=icIRAAAAYAAJ (last access: 20 February 2026), 1865.
Hill, C. A. and Forti, P.: Cave minerals of the World, second edn., National Speleological Society, Huntsville, Alabama, USA, ISBN 1-879961-07-5, 1997.
Kampf, A. R., Roberts, A. C., Venance, K. E., Carbone, C., Belmonte, D., Dunning, G. E., and Walstrom, R. E.: Cerchiaraite-(Fe) and cerchiaraite-(Al), two new barium cyclosilicate chlorides from Italy and California, USA, Mineral. Mag., 77, 69–80, 2013.
Kampf, A. R., Rossman, G. R., Ma, C., Belmonte, D., Biagioni, C., Castellaro, F., and Chiappino, L.: Ramazzoite, [Mg8Cu12(PO4)(CO3)4(OH)24 (H2O)20][(H0.33SO4)3(H2O)36], the first mineral with a polyoxometalate cation, Eur. J. Mineral., 30, 827–834, https://doi.org/10.1127/ejm/2018/0030-2748, 2018.
Kizovski, T. V., Schmidt, M. E., O'Neil, L., Jones, M. W. M., Tosca, N. J., Klevang, D. A., Hurowitz, J. A., Adcock, C. T., Hausrath, E. M., Siebach, K. L., Wolf, Z. U., Sharma, S., VanBommel, S. J., McCubbin, F. M., Cloutis, E., Cable, M. L., Liu, Y., Clark, B. C., Treiman, A. H., Tice, M. M., Catling, D. C., Maki, J., Bosak, T., Weiss, B. P., Fairén, A. G., Christian, J. R., Knight, A. L., Shumway, A. O., Randazzo, N. R., Jørgensen, P. S., Lawson, P. R., Wade, L., Heirwegh, C., Elam, W. T., and Allwood, A. C.: Fe-phosphates in Jezero Crater as evidence for an ancient habitable environment on Mars, Nature Communications, 16, 6470, https://doi.org/10.1038/s41467-025-60026-7, 2025.
Kolitsch, U., Merlino, S., Belmonte, D., Carbone, C., Cabella, R., Lucchetti, G., and Ciriotti, M.E.: Lavinskyite-1M, K(LiCu)Cu6(Si4O11)2(OH)4, the monoclinic MDO equivalent of lavinskyite-2O (formerly lavinskyite), from the Cerchiara manganese mine, Liguria, Italy, Eur. J. Mineral., 30, 811–820, https://doi.org/10.1127/ejm/2018/0030-2731, 2018.
Krivovichev, S. V., Krivovichev, V. G., Hazen, R. M., Aksenov, S. M., Avdontceva, M. S., Banaru, A. M., Gorelova, L. A., Ismagilova, R. M., Kornyakov, I. V., Kuporev, I. V., Morrison, S. M., Panikorovskii, T. L., and Starova, G. L.: Structural and chemical complexity of minerals: An update, Mineralogical Magazine, 86, 183–204, https://doi.org/10.1180/mgm.2022.23, 2022.
La Iglesia, A.: Estimating the thermodynamic properties of phosphate minerals at high and low temperature from the sum of constituent units, Estud. Geol., 65, 109–119, 2009.
Langford, B., Vaks, A., Kutuzov, I., Keinan, J., Golan, T., Zilberman, T., Yasur, G., Morag, N., Ebert, Y., Gaster, O., Ben-Yair, R., Ullman, M., and Frumkin, A.: From hypogenic to biogenic speleogenesis in semi-arid climate: Bat guano-driven carbonate weathering and cave modification in Chariton Cave, Israel. Geomorphology, 483, 109815, https://doi.org/10.1016/j.geomorph.2025.109815, 2025.
Lazaridis, G., Bessas, D., Panou, M., Maróti, B., Crichton, W., Melfos, V., Papadopoulou, L., Gkelis, S., Merkel, D. G., Chumakov, A. I., and Voudouris, P.: Potential biosignatures in goethite deposits in Mavros Vrachos Quarry hypogene cave, CATENA, 242, 108113, https://doi.org/10.1016/j.catena.2024.108113, 2024.
Libowitzky, E.: Correlation of O–H stretching frequencies and O–H...O hydrogen bond lengths in minerals, Monatshefte Für Chemie/Chemical Monthly, 130, 1047–1059, https://doi.org/10.1007/BF03354882, 1999.
Lundberg, J. and McFarlane, D. A.: Post-speleogenetic biogenic modification of Gomantong caves, Sabah, Borneo, Geomorphology, 157, 153–168, 2012.
Lundberg, J. and McFarlane, D. A.: Time-transgressive microbial diversity in a tropical bat guano accumulation, Deer Cave, Mulu, Borneo, Int. J. Speleol., 53, 3, https://doi.org/10.5038/1827-806X.53.3.2508, 2024.
Majchrzak-Kucęba, I. and Nowak, W.: Application of model-free kinetics to the study of dehydration of fly ash-based zeolite, Thermochim. Acta, 413, 23–29, 2004.
Marincea, Ş. and Dumitraş, D.-G.: The occurrence of taranakite in the “dry” Cioclovina Cave (Sureanu Mountains, Romania), Neues Jb. Miner. Monat., 2003, 127–144, https://doi.org/10.1127/0028-3649/2003/2003-0127, 2003.
McFarlane, D. A. and Lundberg, J.: Rates of diagenesis of tropical insectivorous bat guano accumulations: implications for potential paleoenvironmental reconstruction, Int. J. Speleol., 53, 5, https://doi.org/10.5038/1827-806X.53.1.2494, 2024.
Momma, K. and Izumi, F.: VESTA: A Three-Dimensional Visualization System for Electronic and Structural Analysis, J. Appl. Crystallogr., 41, 653–658, https://doi.org/10.1107/S0021889808012016, 2008.
Murray, J. W. and Dietrich, R. V.: Brushite and taranakite from Pig Hole Cave, Giles County, Virginia, Am. Mineral., 41, 616–626, 1956.
Onac, B. P.: Minerals in caves, in Encyclopedia of caves (Third Edition), edited by: White, W. B., Culver, D. C., and Pipan, T., Academic Press, London, United Kingdom, 699–709, https://doi.org/10.1016/B978-0-12-814124-3.00084-4, 2019.
Onac, B. P. and Forti, P.: Minerogenetic mechanisms occurring in the cave environment: an overview, Int. J. Speleol., 40, 79–98, 2011.
Onac, B. P. and Vereş, D. S.: Sequence of secondary phosphates deposition in a karst environment: evidence from Magurici Cave (Romania), Eur. J. Mineral., 15, 741–745, 2003.
Onac, B. P., Breban, R., Kearns, J., and Tǎmaş, T.: Unusual minerals related to phosphate deposits in Cioclovina Cave, Sureanu Mts. (Romania), Theor. Appl. Karstol., 15, 27–34, 2002.
Paragenetic Mode 52 (Mindat): https://www.mindat.org/paragen.php?id=52, last access: 4 June 2025.
Peterson, S. W. and Levy, H. A.: A single-crystal neutron diffraction study of heavy ice, Acta Crystallogr., 10, 70–76, 1957.
Piccini, L., Nannoni, A., Costagliola, P., Paolieri, M., and Vigiani, C.: Composition and structure of phosphate-rich parietal crusts and nodules in Monte Corchia Cave, Alpi Apuane (Central Italy), Front. Earth Sc., 9, 673109, https://doi.org/10.3389/feart.2021.673109, 2021.
Pineau, M., Carter, J., Lagain, A., Ravier, E., Mangold, N., Le Deit, L., Quantin-Nataf, C., and Zanella, A.: Recent aqueous alteration associated to sedimentary volcanism on Mars. Communications Earth & Environment, 6, 800, https://doi.org/10.1038/s43247-025-02713-3, 2025.
Popović, L., de Waal, D., and Boeyens, J. C. A.: Correlation between Raman wavenumbers and P–O bond lengths in crystalline inorganic phosphates, J. Raman Spectrosc., 36, 2–11, 2005.
Postberg, F., Sekine, Y., Klenner, F., Glein, C. R., Zou, Z., Abel, B., Furuya, K., Hiller, J. K., Khawaja, N., Kempf, S., Noelle, L., Saito, T., Schmidt, J., Shibuya, T., Srama, R., and Tan, S.: Detection of phosphates originating from Enceladus's ocean, Nature, 618, 489–493, 2023.
Prasad, P. S. R., Pradhan, A., and Gowd, T. N.: In situ micro-Raman investigation of dehydration mechanism in natural gypsum, Curr. Sci. India, 80, 1203–1207, https://www.currentscience.ac.in/Volumes/80/09/1203.pdf (last access: 20 February 2026). 2001.
Putnis, A.: An Introduction to Mineral Sciences, Cambridge University Press, 480 pp., ISBN 9781139170383, 1992.
Putnis, A., Winkler, B., and Fernandez-Diaz, L.: In situ IR spectroscopic and thermogravimetric study of the dehydration of gypsum, Mineral. Mag., 54, 123–128, 1990.
Queffelec, A., Bertran, P., Bos, T., and Lemée, L.: Mineralogical and organic study of bat and chough guano: implications for guano identification in ancient context, J. Cave Karst Stud., 80, 49–65, 2018.
Redfern, S. A. T.: The kinetics of dehydroxylation of kaolinite, Clay Miner., 22, 447–456, 1987.
Rellini, I., Firpo, M., Martino, G., Riel-Salvatore, J., and Maggi, R.: Climate and environmental changes recognized by micromorphology in Paleolithic deposits at Arene Candide (Liguria, Italy), Quatern. Int., 315, 42–55, 2013.
Robie, R. A., Hemingway, B. S., and Fisher, J. R.: Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 Pascals) pressure and at higher temperatures, Geological Survey Bulletin, 1452, https://doi.org/10.3133/b1452, 1978.
Rodriguez-Navarro, C., Doehne, E., and Sebastian, E.: How does sodium sulfate crystallize? Implications for the decay and testing of building materials, Cem. Concr. Res., 30, 1527–1534, 2000.
Ross, S. D.: Phosphates and other Oxy-anions of Group V, in: The Infrared Spectra of Minerals, edited by: Farmer, V.C., Mineralogical Society of Great Britain and Ireland, London, United Kingdom, 383-422, https://doi.org/10.1180/mono-4.17, 1974.
Rothe, M., Kleeberg, A., and Hupfer, M.: The occurrence, identification and environmental relevance of vivianite in waterlogged soils and aquatic sediments, Earth-Sci. Rev., 158, 51–64, 2016.
Sakae, T. and Sudo, T.: Taranakite from the Onino-Iwaya limestone cave at Hiroshima Prefecture, Japan: A new occurrence, Am. Mineral., 60, 331–334, 1975.
Sauro, F., De Waele, J., Onac, B. P., Galli, E., Dublyansky, Y., Baldoni, E., and Sanna, L.: Hypogenic speleogenesis in quartzite: the case of Corona'e Sa Craba Cave (SW Sardinia, Italy), Geomorphology, 211, 77–88, 2014.
Sendova, M., Hosterman, B. D., and Grebe, A.: In-situ isothermal micro-Raman spectroscopy reveals the activation energy of dehydration in α-FeOOH, J. Raman Spectrosc., 48, 618–622, 2017.
Sergeeva, A. V., Polevoy, L. A., Golikova, M. B., Nazarova, M. A., Gladyshkina, A. V., Kartasheva, E. V., and Kuzmina, A. A.: Spectroscopy of ammonium occupying symmetry-inappropriate positions in crystal structures of salts, J. Appl. Spectrosc., 91, 550–562, 2024.
Shahack-Gross, R., Berna, F., Karkanas, P., and Weiner, S.: Bat guano and preservation of archaeological remains in cave sites, J. Archaeol. Sci., 31, 1259–1272, 2004.
Siljeström, S., Czaja, A. D., Corpolongo, A., Berger, E. L., Li, A. Y., Cardarelli, E., Abbey, W., Asher, S. A., Beegle, L. W., Benison, K. C., Bhartia, R., Bleefeld, B. L., Burton, A. S., Bykov, S. V., Clark, B., DeFlores, L., Ehlmann, B. L., Fornaro, T., Fox, A., Gómez, F., Hand, K., Haney, N. C., Hickman-Lewis, K., Hug, W. F., Imbeah, S., Jakubek, R. S., Kah, L. C., Kivrak, L., Lee, C., Liu, Y., Martínez-Frías, J., McCubbin, F. M., Minitti, M., Moore, K., Morris, R. V., Núñez, J. I., Osterhout, J. T., Phua, Y. Y., Randazzo, N., Hollis, J. R., Rodriguez, C., Roppel, R., Scheller, E. L., Sephton, M., Sharma, S. K., Sharma, S., Steadman, K., Steele, A., Tice, M., Uckert, K., VanBommel, S., Williams, A. J., Williford, K. H., Winchell, K., Wu, M. K., Yanchilina, A., and Zorzano, M.-P.: Evidence of Sulfate-Rich Fluid Alteration in Jezero Crater Floor, Mars, Journal of Geophysical Research: Planets, 129, e2023JE007989, https://doi.org/10.1029/2023JE007989, 2024.
Smith, J. P. and Brown, W. E.: X-ray studies of aluminum and iron phosphates containing potassium or ammonium, Am. Mineral., 44, 138–142, 1959.
Sokol, E. V., Kozlikin, M. B., Kokh, S. N., Nekipelova, A. V., Kulik, N. A., Danilovsky, V. A., Khvorov, P.V., and Shunkov, M. V.: Phosphate record in pleistocene-holocene sediments from Denisova cave: Formation mechanisms and archaeological implications, Minerals, 12, 553, https://doi.org/10.3390/min12050553, 2022.
Soukup, D. A., Buck, B. J., and Harris, W.: Preparing soils for mineralogical analyses. Methods of soil analysis part 5 – mineralogical methods, 5, 13–31, 2008.
Taddei, P., Tinti, A., Bottura, G., and Bertoluzza, A.: Vibrational spectroscopic characterization of new calcium phosphate bioactive coatings, Biopolymers, 57, 140–148, 2000.
Tămaş, T., Miheţ, O., and Giurgiu, A.: Mineralogy of bat guano deposits from Huda lui Papară cave (Trascău Mountains, Romania), Carpath. J. Earth Env., 9, 25–32, 2014.
Vantelon, D., Pelletier, M., Michot, L. J., Barres, O., and Thomas, F.: Fe, Mg and Al distribution in the octahedral sheet of montmorillonites, An infrared study in the OH-bending region, Clay Miner., 36, 369–379, 2001.
Ventruti, G., Ventura, G. D., Bellatreccia, F., Lacalamita, M., and Schingaro, E.: Hydrogen bond system and vibrational spectroscopy of the iron sulfate fibroferrite, Fe(OH)SO4 ⋅ 5H2O, Eur. J. Mineral., 28, 943–952, 2016.
Vieillard, P. and Tardy, Y.: Thermochemical properties of phosphates, in: Phosphate Minerals, edited by: Nriagu, J. O., and Moore, P. B., Springer-Verlag Berlin, Heidelberg, Germany, 171–198, https://doi.org/10.1007/978-3-642-61736-2_4, 1984.
Wu, Y., Xiao, Z., Wu, Y., Pan, L., Yan, P., Liao, S., Pan, Q., Li, S., Li, Y., and Hsu, W.: Hydrothermal fluid activity on Mars recorded in phosphates of the gabbroic shergottite Northwest Africa 13581. J. Geophys. Res-Planet., 129, e2024JE008412, https://doi.org/10.1029/2024JE008412, 2024.
Zabat, M. and Van Damme, H.: Evaluation of the energy barrier for dehydration of homoionic (Li, Na, Cs, Mg, Ca, Ba, Alx(OH and La)-montmorillonite by a differentiation method, Clay Miner., 35, 357–363, 2000.