the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Feb 2020
| 11 Feb 2020
Breyite inclusions in diamond: experimental evidence for possible dual origin
Alan B. Woodland
Andrei V. Girnis
Vadim K. Bulatov
Gerhard P. Brey
Heidi E. Höfer
Inclusions of breyite (previously known as walstromite-structured CaSiO3) in diamond are usually interpreted as retrogressed CaSiO3 perovskite trapped in the transition zone or the lower mantle. However, the thermodynamic stability field of breyite does not preclude its crystallization together with diamond under upper-mantle conditions (6–10 GPa). The possibility of breyite forming in subducted sedimentary material through the reaction CaCO3 + SiO2 = CaSiO3 + C + O2 was experimentally evaluated in the CaO–SiO2–C–O2 ± H2O system at 6–10 GPa, 900–1500 ∘C and oxygen fugacity 0.5–1.0 log units below the Fe–FeO (IW) buffer. One experimental series was conducted in the anhydrous subsystem and aimed at determining the melting temperature of the aragonite–coesite (or stishovite) assemblage. It was found that melting occurs at a lower temperature (∼1500 ∘C) than the decarbonation reaction, which indicates that breyite cannot be formed from aragonite and silica under anhydrous conditions and an oxygen fugacity above IW – 1. In the second experimental series, we investigated partial melting of an aragonite–coesite mixture under hydrous conditions at the same pressures and redox conditions. The melting temperature in the presence of water decreased strongly (to 900–1200 ∘C), and the melt had a hydrous silicate composition. The reduction of melt resulted in graphite crystallization in equilibrium with titanite-structured CaSi2O5 and breyite at ∼1000 ∘C. The maximum pressure of possible breyite formation is limited by the reaction CaSiO3 + SiO2 = CaSi2O5 at ∼8 GPa. Based on the experimental results, it is concluded that breyite inclusions found in natural diamond may be formed from an aragonite–coesite assemblage or carbonate melt at 6–8 GPa via reduction at high water activity.
- Article
(13416 KB) - Full-text XML
- BibTeX
- EndNote





Since the discovery by Joswig et al. (1999), CaSiO3 inclusions in diamond have been reported by a number of researchers. Monomineralic CaSiO3 inclusions as well as CaSiO3 coexisting with larnite, titanite-structured CaSi2O5, calcite (aragonite), perovskite, silica polymorphs, orthopyroxene (possibly converted from bridgmanite), clinopyroxene, ringwoodite, ferropericlase, monticellite and cuspidine have been described in diamonds from Kankan, Guinea (Joswig et al., 1999; Stachel et al., 2000; Nasdala et al., 2003); Juína, Brazil (Hayman et al., 2005; Brenker et al., 2007; Walter et al., 2008; Wirth et al., 2009; Bulanova et al., 2010; Pearson et al., 2014; Anzolini et al., 2016; Kaminsky et al., 2016); Machado River, Brazil (Burnham et al., 2016); and the Slave province, Canada (Davies et al., 2004; Tappert et al., 2005). CaSiO3 was also documented as a daughter mineral of multiphase inclusions in Juína diamond, which presumably formed through the crystallization of carbonatitic melts (Kaminsky et al., 2009). Most of the CaSiO3 phases were identified as triclinic wollastonite II or walstromite-structured CaSiO3. The two polymorphs have very similar crystal structures (Joswig et al., 2003), but their relative stability relations are unknown (Dörsam et al., 2009). Therefore, the terms wollastonite II and walstromite-structured CaSiO3 have often been used synonymously. The name breyite was recently proposed for the latter phase and approved by IMA in 2018 (IMA no. 2018-062; Brenker et al., 2018).
Calcium metasilicate CaSiO3 occurs in a number of structural modifications, and its phase diagram has been extensively studied. Ringwood and Major (1967) converted wollastonite to a denser phase (breyite) at 3 GPa and ∼900 ∘C. The wollastonite–breyite boundary was then tightly constrained by experiments (e.g., Essene, 1974; Gasparik et al., 1994; Akaogi et al., 2004). Kanzaki et al. (1991) observed breyite decomposition to larnite (Ca2SiO4) and titanite-structured CaSi2O5 at 12 GPa and 1500 ∘C. This reaction was studied in detail by Kubo et al. (1997) and Akaogi et al. (2004). Sueda et al. (2006) experimentally demonstrated that the larnite + CaSi2O5 assemblage transforms to CaSiO3 perovskite at a pressure of ∼14 GPa. Gasparik et al. (1994) determined the melting temperatures of CaSiO3 polymorphs at pressures of 8–16 GPa. Phase relations of CaSiO3 are summarized in Fig. 1.
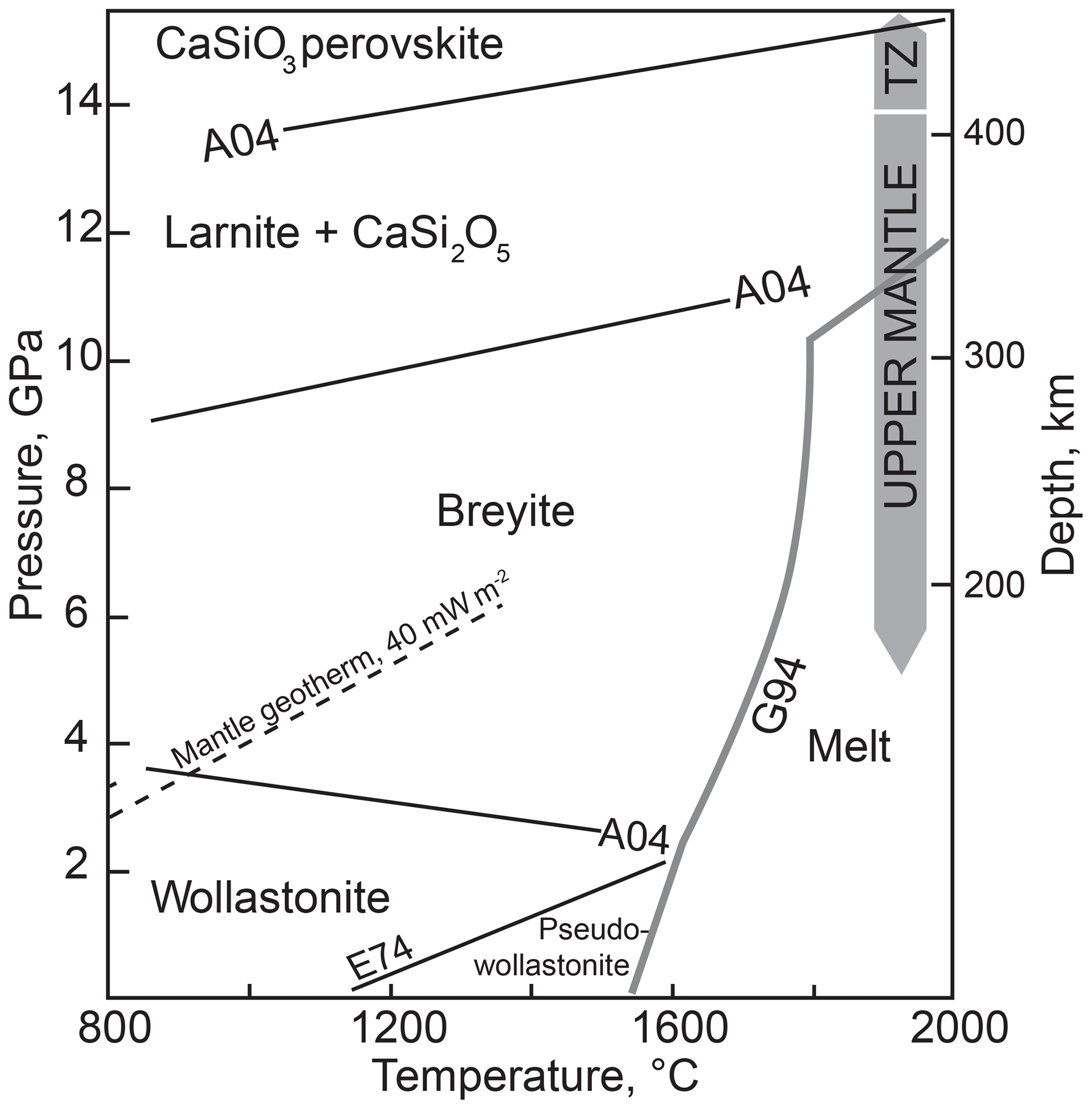
Figure 1Schematic phase diagram of CaSiO3 under moderate pressure–temperature conditions. Sources of data: A04, Akaogi et al. (2004); E74, Essene (1974); G94, Gasparik et al. (1994). TZ is the transition zone. The conductive mantle geotherm for a heat flow of 40 mW m−2 is shown following Hasterok and Chapman (2011). The field of monoclinic wollastonite II (Chatterjee et al., 1984) is omitted.
It is clear from the phase diagram (Fig. 1) that the interpretation of monomineralic breyite inclusions in diamond may be ambiguous. The more common scenario implies the entrapment of CaSiO3 perovskite in the transition zone or lower mantle and its conversion to breyite upon decompression (e.g., Joswig et al., 1999; Brenker et al., 2007; Harte, 2010; Kaminsky, 2012; Burnham et al., 2016). This interpretation is favored by the ubiquity of CaSiO3 perovskite in the lower mantle (e.g., Wood, 2000; Irifune and Tsuchiya, 2007), the occurrence of breyite together with ferropericlase ± bridgmanite in a single diamond (Stachel et al., 2000) and coexistence of breyite with ringwoodite in a diamond (Pearson et al., 2014). One problem is the enormous volume change of the CaSiO3 perovskite to breyite transformation of ∼38 % (Akaogi et al., 2004). Anzolini et al. (2016) estimated a minimum entrapment pressure of breyite in Juína diamond as 5.7 GPa at 1500 K and argued that the inclusion could not have converted from CaSiO3 perovskite because of the large volume change involved. To accommodate such a volume change, diamond had to have experienced cracking or considerable plastic deformation, features not detected by Anzolini et al. (2016). In contrast, plastic and brittle deformations were observed around breyite inclusions in diamond by Cayzer et al. (2008), Zedgenizov et al. (2015), Burnham et al. (2015, 2016) and Anzolini et al. (2018), which emphasizes the possibility of the dual origin of breyite inclusions in diamond.
The suggestion that a breyite inclusion can form through direct entrapment at 4–10 GPa rather than from a perovskite precursor is consistent with the CaSiO3 phase diagram (Fig. 1). In this study we explore this possibility by high-pressure experiments in the simple CaO–SiO2–C–O2 system with or without H2O.
In contrast to CaSiO3 perovskite, breyite is not stable in a peridotitic mantle because of the reaction with low-Ca pyroxene:
High-Ca pyroxene is stable up to ∼17–18 GPa (Akaogi et al., 2004), i.e., above the breyite stability field. There are no CaSiO3 phases in subducted basaltic crust (MORB) up to ∼20 GPa (e.g., Perrillat et al., 2006). Hence, another lithology for breyite crystallization must be looked for. Wollastonite occurs very rarely in high-pressure rocks (Chopin et al., 2008; Galvez et al., 2013a, b) and is absent in subducted sediments (Li and Schoonmaker, 2003). However, the association of calcite and silica is very common in oceanic sediments (Li and Schoonmaker, 2003) and could be a viable source of CaSiO3 formation via the decarbonation reaction
Hence, the system CaO–SiO2–C–O2 rather than CaSiO3–C should be considered. Litasov et al. (2017) calculated the position of Reaction (R2) at high pressures under fluid-saturated conditions (Fig. 2). They showed that Reaction (R2) intersects the melting curve of CaCO3 at pressure close to the graphite–diamond transformation. However, Reaction (R2) becomes metastable even at significantly lower pressures because of the occurrence of the melting reaction
as indicated by experiments in the CaO–SiO2–CO2 system by Wyllie and Huang (1976) and Huang et al. (1980). These studies showed that Reaction (R2) terminates in an invariant point at ∼1.8 GPa and 1320 ∘C and is metastable at higher pressures. The position of Reaction (R3) in the diamond stability field was not studied experimentally, but it is clear that breyite cannot be produced by decarbonation Reaction (R2) in the presence of a CO2 fluid or, more precisely, a CO2–CO fluid whose composition depends on oxygen fugacity (fO2).
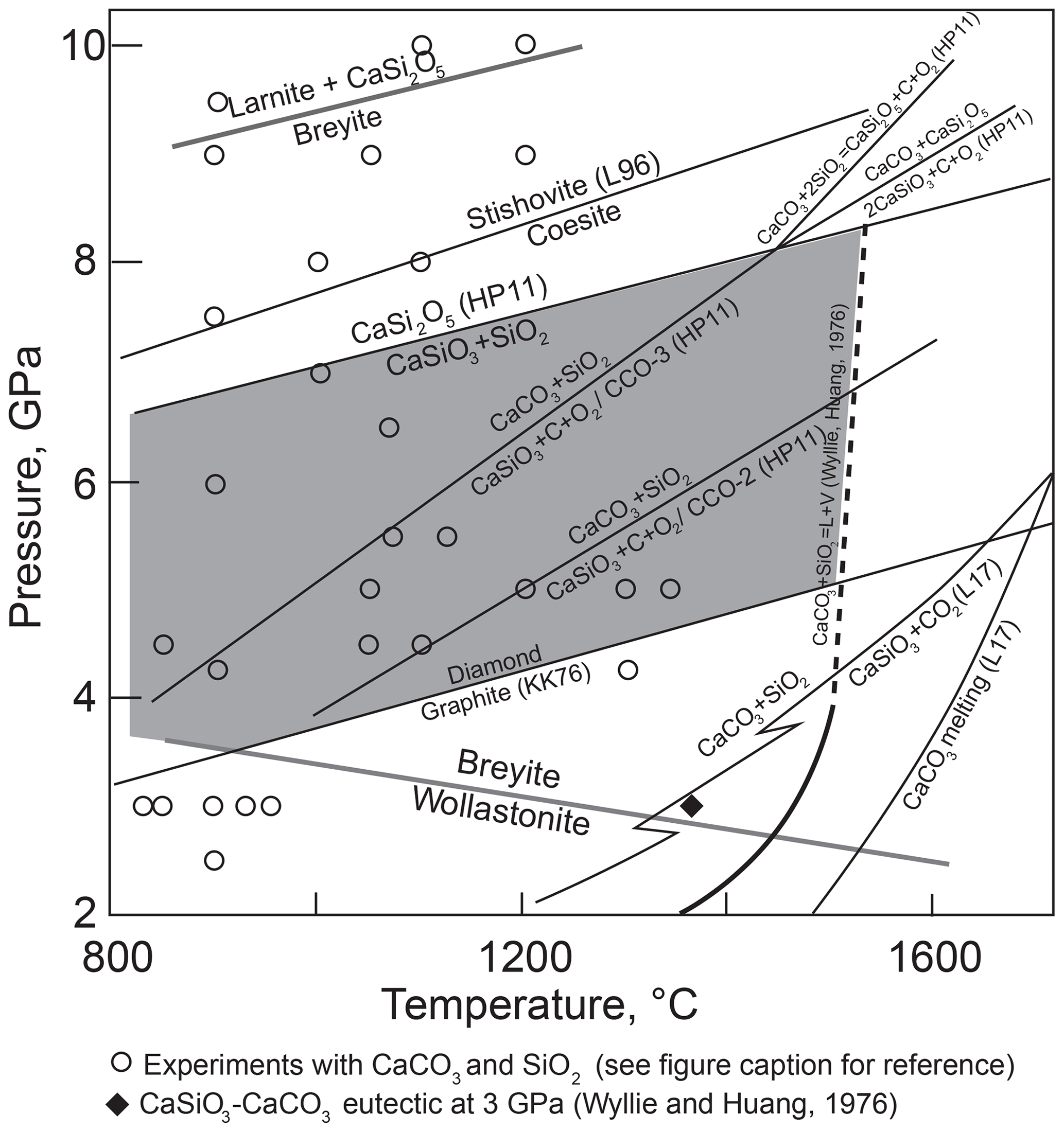
Figure 2Selected equilibria of the CaO–SiO2–C–O2 system relevant for the conditions of breyite crystallization via decarbonation Reaction (R2). Solid gray lines show CaSiO3 transformation reactions wollastonite to breyite and breyite to larnite + CaSi2O5 (Fig. 1). Other lines show experimental and theoretical estimates of equilibria in the system. Sources of data: L96, Liu et al. (1996); HP11, Holland and Powell (2011); L17, Litasov et al. (2017); KK, Kennedy and Kennedy (1976). The reaction CaCO3 + SiO2 = CaSiO3 + CO2 was calculated by Litasov et al. (2017) for fluid-saturated conditions; kinks on the reaction line are related to the quartz–coesite and wollastonite–breyite transformations. The reaction lines CaCO3 + SiO2 = CaSiO3 + C + O2 were calculated using thermodynamic data from Holland and Powell (2011) for logfO2 values CCO – 2 and CCO – 3 in the absence of fluid. Melting of CaCO3 + SiO2 was experimentally studied up to 3 GPa (Wyllie and Huang, 1976; Huang et al., 1980) and is extrapolated arbitrarily to higher pressures as a dashed line. Circles show melting experiments in hydrous and anhydrous systems in which coexisting CaCO3 and SiO2 were observed (Hammouda, 2003; Yaxley and Brey, 2004; Grassi and Schmidt, 2011a, b; Tsuno and Dasgupta, 2012; Kiseeva et al., 2012, 2013; Poli, 2015; Brey et al., 2015; Mann and Schmidt, 2015; Martin and Hermann, 2018). The shaded field shows the conditions of possible formation of breyite inclusions in diamond.
Decarbonation is possible at lower temperatures, if CO2 activity is lower than that in the presence of CO2 fluid or, alternatively, at a fO2 below the C–CO–CO2 buffer, via the reaction
Another reaction limiting the possible conditions of breyite crystallization in the diamond stability field is
Experiments in the CaSiO3–CaTiO3 (Kubo et al., 1997) and CaTiSiO5–CaSi2O5 systems (Knoche et al., 1998) at 5.3–16 GPa and 1200 and 1350 ∘C, respectively, showed that the breyite–coesite assemblage transforms to CaSi2O5 in the Ti-free system between 5.3 and 8.7 GPa. Calculations using the thermodynamic data of Holland and Powell (2011) indicate that equilibrium (Reaction R5) lies ∼3.0 GPa above the graphite–diamond boundary (Fig. 2). It is evident that Reaction (R5) limits the pressure range where breyite can form in metasedimentary (silica-saturated) assemblages.
With these considerations in mind, our experimental study had the following goals:
-
to constrain the temperature dependence of melting Reaction (R3) at 6–10 GPa and its position relative to redox Reaction (R4) at low but geologically realistic oxygen fugacities near the iron–wüstite (IW) buffer,
-
to model the formation of CaSiO3 at low CO2 activity in the presence of mixed C–O–H fluid, and
-
to obtain experimental constraints for the lower stability limit of Reaction (R5).
Two types of experiments were conducted. (1) One configuration consisted of three layers (from bottom to top): Fe metal, SiO2 and CaCO3. These experiments simulated melting of CaCO3 (aragonite) + SiO2 (coesite or stishovite) under anhydrous conditions with simultaneous carbonate reduction at a fO2 near IW. (2) The second configuration comprised two layers, a mixture of Fe metal and Mg(OH)2 in the lower part of the sample and a SiO2–CaCO3 mixture in the upper part, nonhermetically separated by a Pt disc. These experiments simulated the interaction of CaCO3 and SiO2 with a mixed reduced fluid produced by the reaction
and subsequent interaction of H-bearing species with CaCO3 in the upper zone.
Starting materials for the experiments were prepared from Fe, SiO2 and Mg(OH)2 reagents along with natural calcite (Icelandic spar). The initial components were pulverized in an agate mortar to a grain size <20 µm and mixed, when necessary, by grinding under ethanol. Sample powders were packed into Pt capsules with a 1.4 mm inner diameter and ∼2.5 mm long.
The experiments were performed in a Walker-type multianvil apparatus using tungsten carbide cubes with a truncation edge length of 8 mm. The pressure assembly consisted of a 95 % MgO + 5 % Cr2O3 octahedron, a zirconia sleeve, a Re foil heater and MgO inserts around the capsule. Any free space was filled with Al2O3-based cement of W. Haldenwager Technische Keramic GmbH. Pressure was calibrated against the Mg2SiO4 olivine–wadsleyite phase transition at 1200 ∘C and 13.6 GPa (Morishima et al., 1994), the coesite–stishovite transition at 1650 ∘C and 10.6 GPa (Zhang et al., 1996), and the CaGeO3 garnet–perovskite transition at 1200 ∘C and 5.6 GPa (Ross et al., 1986). The experimental apparatus and pressure calibration were described in detail by Brey et al. (2008). The uncertainty in pressure was estimated as ±0.3 GPa. Temperature was measured with a W95Re5–W74Re26 thermocouple inserted through an Al2O3 ceramic tube at the top end of the capsule. Temperature fluctuations during the experiments were no higher than ±3 ∘C. The duration of the experiments was 1 to 45 h (Table 1). The experiments were terminated by shutting off the power supply. After the experiment, the capsule was mounted into epoxy, sectioned lengthwise and polished with oil-based diamond pastes. The products were analyzed with a Jeol 8900 Superprobe electron microprobe in wavelength dispersive mode. The microprobe is equipped with five spectrometers along with an energy-dispersive spectroscopy (EDS) system. The following natural and synthetic phases were used as standards: wollastonite for Si and Ca, Al2O3 for Al, fayalite for Fe, forsterite for Mg, and albite for Na. The analytical conditions were as follows: 15 kV accelerating voltage, 20 nA beam current, and 20–40 s counting time for peak and background. Minerals were analyzed with a focused beam and quenched melts, with a beam defocused to a 20 µm spot size. Some experiments were analyzed with a Jeol JSM-6490 scanning electron microscope equipped with an INCA analytical system. The accuracy of INCA EDS analysis was inferior to that of the electron microprobe, and these analyses were considered semiquantitative. However, the quality of these analyses was sufficient for identification of experimental phases, most of which were of fixed composition.
Table 1Conditions and products of experiments.
Mineral abbreviation: Arg, aragonite; Br, breyite; Coe, coesite; Coh, cohenite; Di, diopside; En, enstatite; Fa, fayalite; Fo, forsterite; FPer, ferropericlase; Fs, ferrosilite; Gr, graphite; Ol, Fe–Mg olivine; Opx, Fe–Mg pyroxene; Sti, stishovite
In all anhydrous experiments (type 1), metallic iron coexisted with wüstite (pure FeO), which indicates that the fO2 was close to the IW buffer reaction:
However, careful inspection of the experimental products revealed that the actual fO2 could differ from IW. In most cases, the association of metallic iron and wüstite was observed in the central part of the metal zone, whereas its outer part consisted only of pure iron, suggesting that the fO2 was below IW. Some oxygen fugacity gradient existed evidently between the inner and outer parts of the metal zone. It is reasonable to suggest that the oxygen fugacity in the carbonate zone was controlled by equilibria at the interface between the iron and silica zones. Ferrosilite and fayalite were observed in most experiments of this series between iron metal and silica. Hence, the oxygen fugacity was controlled by the reactions
or, when fayalite was absent,
Reactions (R8) and (R9) are sums of equilibrium (Reaction R7) and the following reactions:
respectively. Therefore, the logfO2 relative to the IW buffer can be calculated as
for the fayalite–ferrosilite–iron assemblage and
for the iron–ferrosilite–silica (coesite or stishovite), where ΔG(i) is the Gibbs free energy change of the respective reaction, T is the absolute temperature and R is the universal gas constant. The fO2 was calculated using the thermodynamic properties of phases from the dataset of Holland and Powell (2011). In most cases, the resulting fO2 values lie 0.5–1.0 log units below IW (Table 1). To further decrease the fO2 in one experiment (M753), San Carlos olivine (Mg no. ∼0.9) was added to the starting Fe layer. This produced Fe–Mg olivine and orthopyroxene solid solutions in the Fe zone, which imposed a fO2 of ∼IW – 1.

Figure 3Back-scattered electron (BSE) images of the products of anhydrous melting experiment M751 (8 GPa, 1500 ∘C). (a) Overview of the sample capsule. Black material outside the Pt capsule is epoxy. (b–d) Details of the sample indicated by rectangles in (a). Mineral symbols: Arg, aragonite; Coe, coesite; Fa, fayalite; and Fs, ferrosilite.
In the hydrous experiments (type 2), the buffer assemblage consisted always of metallic Fe free of measurable Pt or any other admixture and ferropericlase with of approximately 0.3–0.4 (Table 1). The composition of ferropericlase was rather uniform in all experiments, with no systematic variations occurring in contact with the capsule. For these experiments, the fO2 relative to IW was calculated as , where aFeO is the activity of FeO in ferropericlase, calculated using the model of O'Neill et al. (2003). The resulting fO2 values are lower than IW by 0.2–0.3 log units.
4.1 Anhydrous aragonite–SiO2 melting
In the subsolidus experiments at temperatures ≤1300 ∘C, the three layers remained separated with only minor interaction between Fe metal and SiO2 that produced ferrosilite and fayalite, whereas no interaction was observed between aragonite and SiO2. At higher temperatures, quenched melt appeared at the contacts between aragonite and SiO2 (Fig. 3). The first melt portion appears near the walls of capsules, in the zones of highest temperature. The amount of melt increases rapidly between 1400 and 1500 ∘C (Fig. 4). Minor graphite grains were observed in some experiments in or near the melt pools. Their occasional presence within carbonate crystals suggests that graphite was present during the experiment and is not a quench phase. In the most reduced experiment with olivine in the Fe zone (M753 at 8 GPa, 1300 ∘C and logfO2 ∼IW – 1), more extensive graphite formation was observed (Fig. 5). In this experiment, graphite formed a continuous band of small crystals near the coesite–aragonite contact. Despite the considerable amount of graphite, breyite was not observed in this run, indicating that Reaction (R2) remains metastable under such conditions.
4.2 Hydrous experiments
Melt was always present as a major phase in these experiments. The quench texture was very different from that observed in the anhydrous experiments and consisted of a very fine-grained or even glassy matrix and acicular quench coesite or stishovite crystals (Fig. 6). Equilibrium growth of crystals often occurred along the walls or bottom of the capsule. CaSi2O5 formed large euhedral crystals up to 30–50 µm across (Fig. 6a, b). Stishovite crystallized as long prismatic crystals, which were easily distinguished from isometric coesite grains. Breyite was observed in minor amounts at 6–8 GPa (Fig. 7) as short-to-long prismatic grains up to 20 µm long (Fig. 6b–d). Diopside occurred in some of the experiments due to Mg contamination from the lower buffer zone. Graphite was present in all experiments both within the melt phase and as inclusions in silicate crystals.
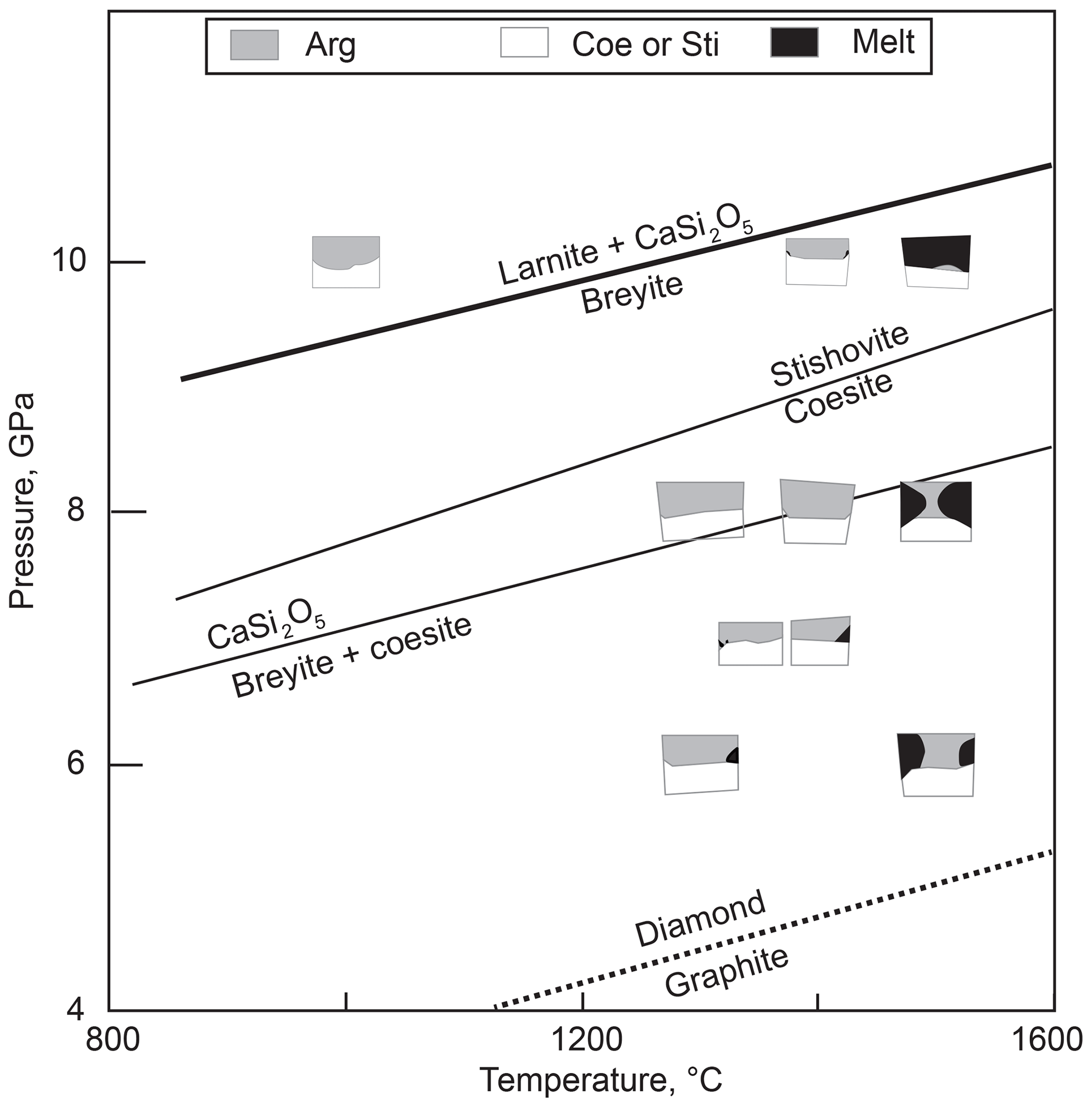
Figure 4Summary of anhydrous melting experiments. Symbols show schematic relations of run products. Sources of data for mineral reactions are the same as in Figs. 1 and 2.
4.3 Phase compositions
The solid phases mostly have constant compositions and approach ideal stoichiometric element ratios (Table 2). Through contamination with the buffer materials, minor amounts of Mg and Fe were occasionally present in the calcium silicate phases. Aragonite and silica minerals were almost always free of Fe and Mg.
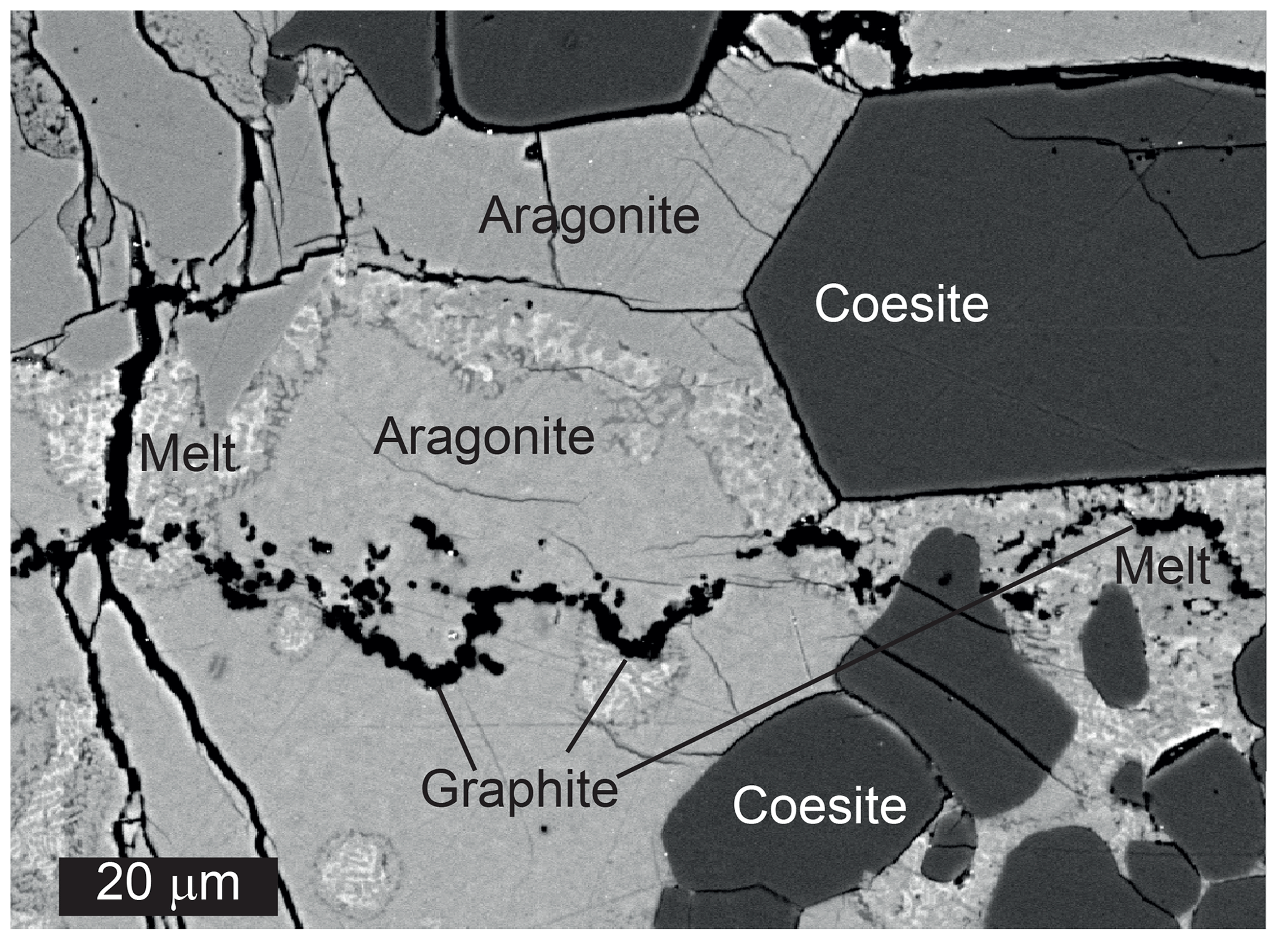
Figure 5BSE illustrating simultaneous melting and reduction near the aragonite–coesite contact in run M753 at 8 GPa, 1300 ∘C and at IW – 1. This run was conducted under more reduced conditions compared with other experiment, which resulted in more extensive graphite formation. Note that, despite significant reduction, breyite was not formed. The melt zone apparently moved upward, probably under the influence of a temperature gradient.
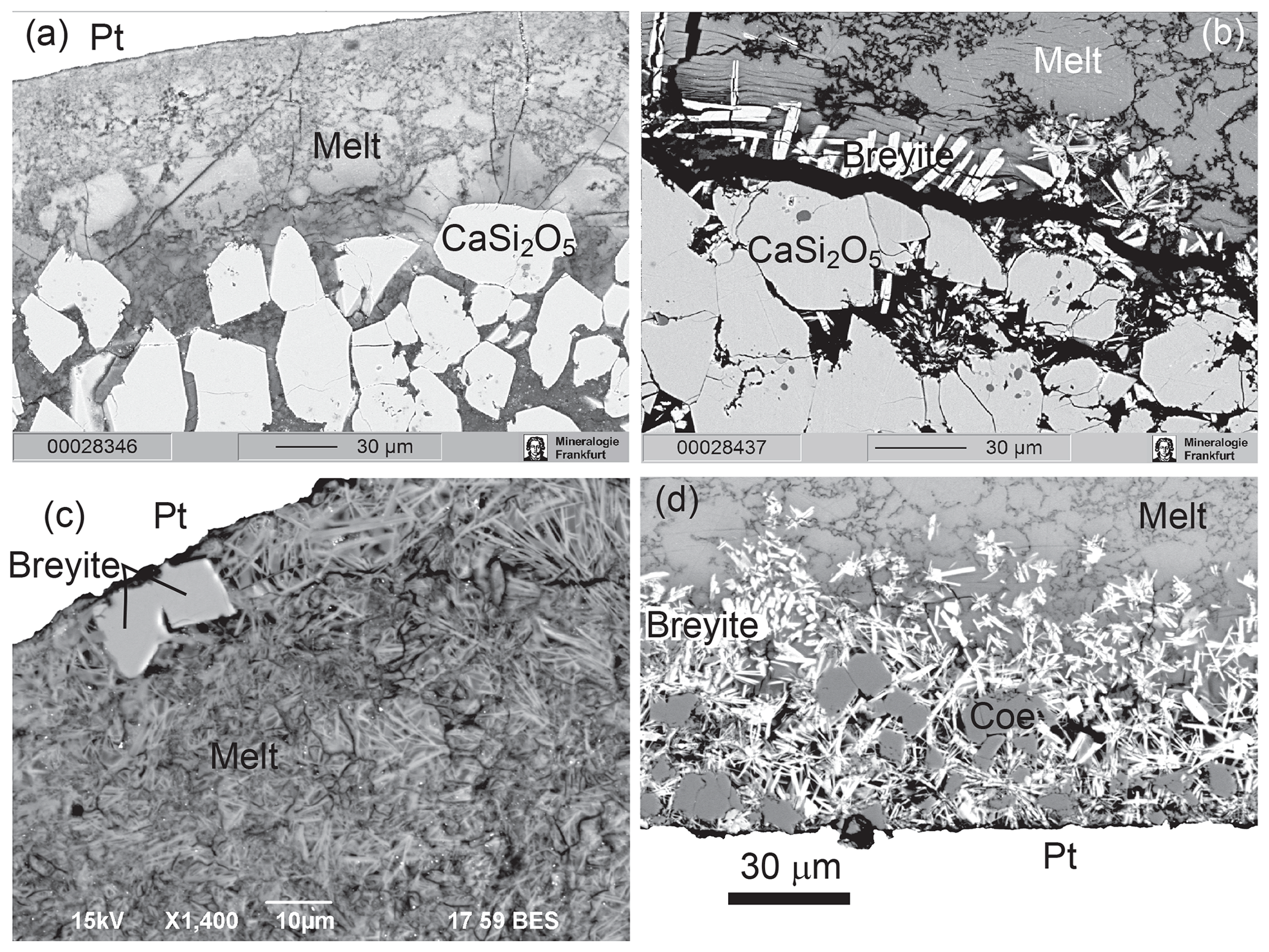
Figure 6BSE images of hydrous experiments. (a) Run M745 (10 GPa, 1000 ∘C). Euhedral crystals of CaSi2O5 in the lower part of the sample below the zone of pure melt. (b) Run M747 (8 GPa, 1000 ∘C). Breyite crystallization between the layer of CaSi2O5 grains and melt. The black band between breyite and CaSi2O5 is a crack filled with epoxy. (c) Run M775 (6 GPa, 1000 ∘C). Breyite crystals near the wall of the Pt capsule in quenched melt. (d) Run M778 (6 GPa, 900 ∘C). Coesite grains and long prismatic to acicular breyite crystals at the bottom of the sample.
Table 2Compositions of experimental phases, wt %.

One standard deviation is given in parentheses.
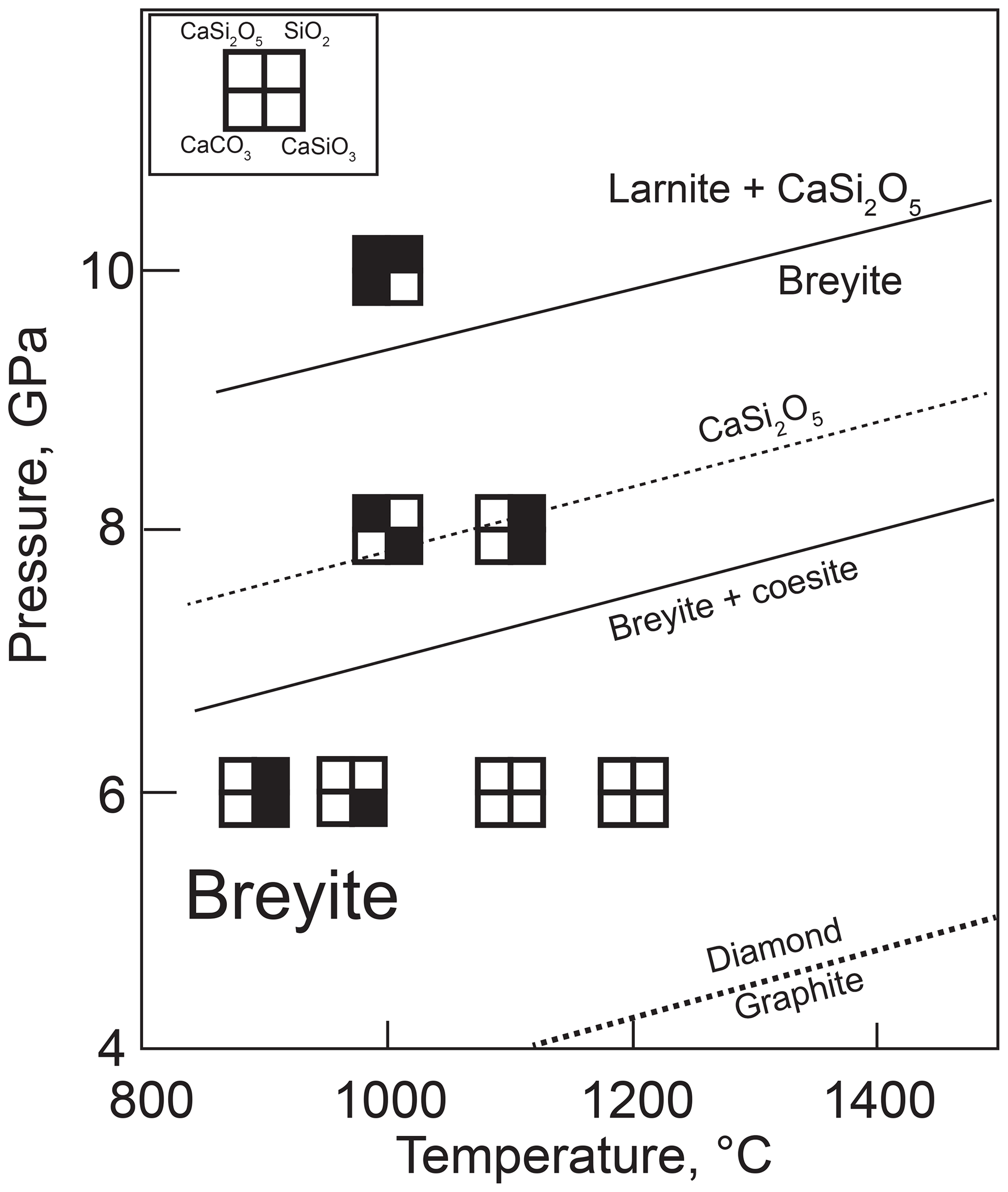
Figure 7Summary of hydrous melting experiments. Sources of data for mineral reactions are the same as in Figs. 1 and 2. The position of the reaction breyite + coesite = CaSi2O5 is shown as calculated from the thermodynamic data of Holland and Powell (2011) (solid line) and from the results of our experiments (dashed line). Note that an additional melt phase is present in all experiments.

Figure 8Silica contents in quenched melts from the anhydrous and hydrous experiments. Melts from the anhydrous experiments labeled “+FeO” and “+MgO” were contaminated with FeO and MgO, respectively, from buffer mixtures at a level of several weight percent (Table 2).
The melt composition from the anhydrous melting experiments was rather variable, especially at low temperatures and low melt fractions (Table 2, Fig. 8). This is related to contamination of the CaCO3–SiO2 system by Fe from the buffer mixture and, possibly, other chemical components from the starting reagents. The influence of these components was most significant at low melt fractions, as indicated by significant FeO and MgO contents in the melt from some experiments. Melt from run M770 (6 GPa, 1300 ∘C) contained up to 18 wt % FeO, and its SiO2 content was also unusually high, ∼30 wt %. At higher temperatures, the amount of impurities decreased and the melt shifted to a Ca carbonate composition with 5–10 wt % SiO2 (Fig. 8). The melt composition is essentially insensitive to temperature above 1300 ∘C. The SiO2 content seems to decrease with increasing pressure, although the available data are insufficient for a quantitative assessment.
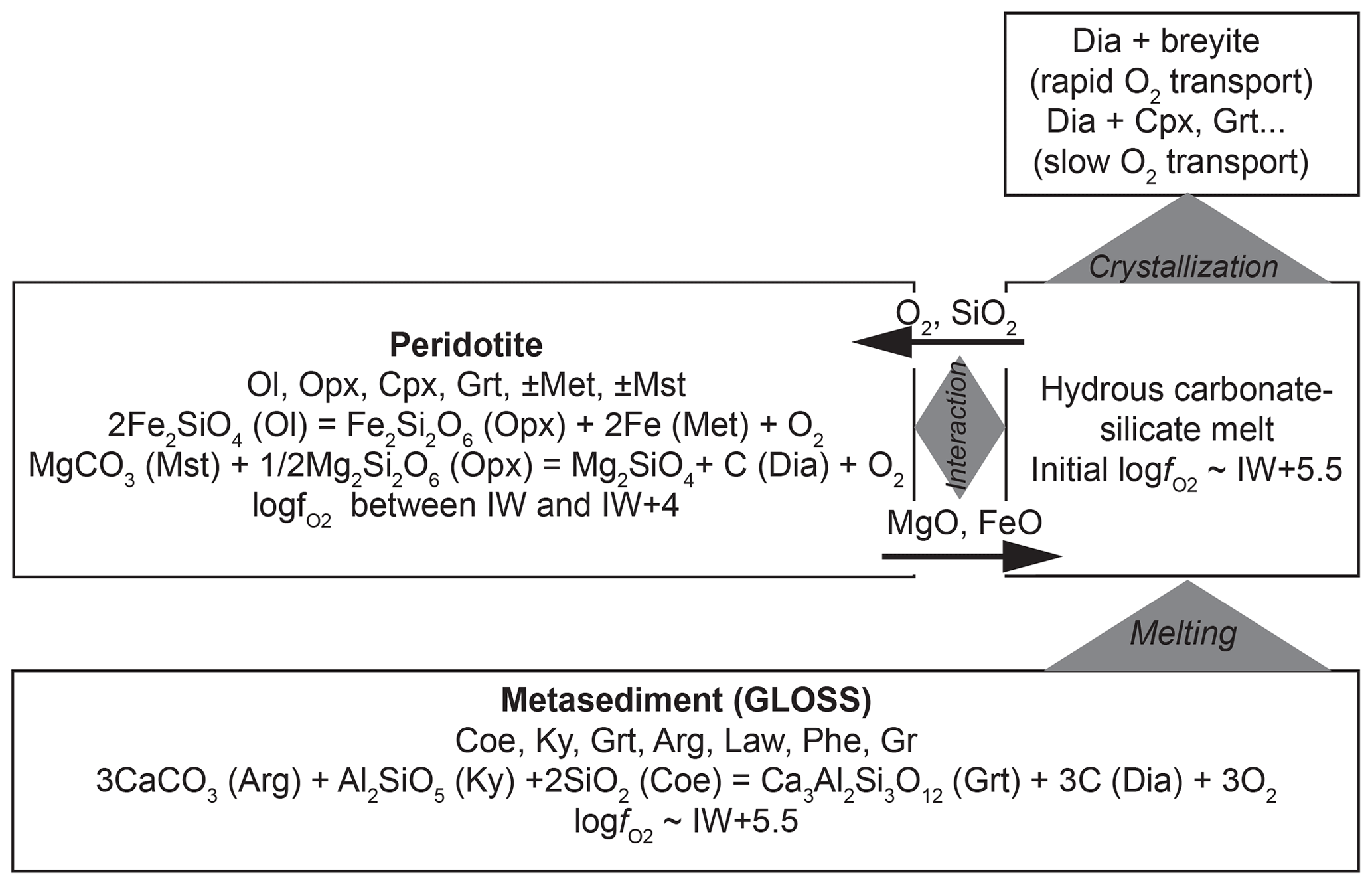
Figure 9Flow diagram describing a possible scenario for the formation of diamond-hosted breyite inclusions at upper-mantle conditions (6–8 GPa and 1000 ∘C) through interaction of hydrous carbonated metapelite and reduced peridotite. Mineral assemblages and reactions controlling fO2 are shown. Black arrows show the fluxes of main components under the influence of chemical potential gradients (e.g., Bulatov et al., 2014).
The compositions of melts from the hydrous experiments are different and can be qualified as hydrous calcium silicate liquids. The content of carbonate species is probably low, which is suggested by the absence of quench carbonates and the abundance of graphite produced by the reduction of starting CaCO3. The SiO2 content ranges from ∼35 to 55 wt % and is insensitive to temperature but decreases with increasing pressure (Fig. 8).
5.1 Decarbonation versus melting reactions
The position of melting Reaction (R3) can be only approximately constrained by our experiments. Its eutectic nature is supported by the absence of additional phases over the melting interval and the independence of melt composition on the degree of melting at high temperatures. However, there are obviously departures from the eutectic behavior. The solid assemblage is transformed to melt (until exhaustion of either silica of aragonite) over a considerable temperature interval rather than at a constant temperature (Fig. 4). There are several reasons for such behavior. First, there is a temperature gradient within the capsule. Our estimates (Woodland et al., 2018) indicate that temperature variation within the short capsules used in the experiments could be up to 50 ∘C. This explains the gradual propagation of the melt zone from capsule walls toward the center of the sample. Secondly, Fe contamination was observed in many experiments, and significant amounts of FeO were dissolved in melts at low melt fraction. This explains the appearance of small amounts of melt very far from the liquidus and the slow increase in the degree of melting at temperatures 100–200 ∘C below the liquidus. Thirdly, the presence of minor amounts of graphite in some experiments indicates that the melting was at least not always eutectic but peritectic in nature via the reaction
Finally, the presence of minor amounts of H2O and/or CH4 cannot be excluded because of impurities in the starting materials and possible hydrogen migration into the capsule during the experiment. Hence, the melt composition was not constant but changed with increasing temperature, causing melting to occur over a finite temperature interval. However, at high melt fractions, the influence of contamination decreases, and the position of disappearance of aragonite should be close to the position of the eutectic reaction.
Our results suggest that Reaction (R3) occurs at temperatures slightly higher than 1500 ∘C at 6–8 GPa and at ∼1500 ∘C at 10 GPa. The increase in melt fraction as pressure increases from 6 to 8 GPa at 1500 ∘C suggests that Reaction (R3) could have a negative slope in the coesite stability field. This is in line with a very weak to negligible pressure effect on the melting temperature of CaCO3 above 5 GPa (Li et al., 2017). At 10 GPa, stishovite occurs instead of coesite, which results in a larger volume change of the melting reaction and a different (though unconstrained) slope of this melting reaction.
The absence of CaSiO3 in all experiments of this series implies that Reaction (2) is metastable under CO2-undersaturated conditions at a fO2≈ IW – 0.5. This result is at odds with calculations of Reaction (R2) using the thermodynamic properties of phases from Holland and Powell (2011). These calculations indicate that a decrease in fCO2 and, correspondingly, fO2 shifts Reaction (R2) to lower temperatures. As depicted in Fig. 2, already at fO2 values 3 log units below the C–CO–CO2 (CCO) equilibrium (0.5–3.0 log units above the IW buffer), Reaction (R2) occurs at lower temperatures than CaCO3–SiO2 melting over the whole pressure range of breyite stability. Hence, breyite should have been stable rather than aragonite and coesite at least in our highest temperature experiments. The absence of CaSiO3 in the experimental products could be related to the low melting temperature of the CaSiO3 + CaCO3 assemblage. At 3 GPa, the eutectic melting of this assemblage occurs ∼100 ∘C below the CaCO3 + SiO2 eutectic melting point (Huang and Wyllie, 1974). The melting relations of CaSiO3 + CaCO3 at 6 GPa were recently studied by Fedoraeva et al. (2019). They determined the aragonite–breyite eutectic temperature to be 1400 ∘C at 6 GPa, which is lower than the temperature of melting Reactions (R3) and (R12) at this pressure.
The metastability of the aragonite–coesite–breyite assemblage is indirectly supported by the results of other experimental studies. Figure 2 shows the pressure–temperature conditions of melting experiments in various systems in which silica and aragonite (calcite) are present. The silica–calcite–wollastonite assemblage was observed in a single experiment on calcite assimilation in dacitic melt at 0.5 GPa and 1000 ∘C (Carter and Dasgupta, 2016). In all previous experiments above 3 GPa, the CaCO3 + SiO2 assemblage remained stable up to 1400 ∘C under uncontrolled fO2. Thus, we conclude that at a fO2 0.5–1.0 log units below IW, decarbonation Reaction (R2) is metastable owing to the occurrence of eutectic melting.
5.2 The equilibrium CaSiO3 + SiO2 = CaSi2O5
Equilibrium (Reaction R5) additionally limits the conditions of possible breyite formation from CaCO3 and SiO2. Although it has not been systematically studied experimentally, the position of this equilibrium in pressure–temperature space can be estimated from the thermodynamic data of Holland and Powell (2011). For example, CaSi2O5 should become stable at 1100 ∘C at a pressure of 7.3 GPa. As plotted in Fig. 2, it is apparent that the stability field of breyite is reduced by ∼2 GPa if the metasedimentary assemblage is SiO2-saturated. However, the results of our hydrous experiments can provide some additional constraints on the actual position of this reaction. Of three experiments performed at 8 GPa and 1100 ∘C, one produced CaSi2O5 (Table 1, Fig. 7). The other two experiments at this temperature produced either breyite + SiO2 or only breyite. In an additional experiment at 1000 ∘C, CaSi2O5 was found to coexist with breyite but without coesite (Table 1). Thus, the equilibrium (Reaction R5) must lie very close to 8 GPa at 1100 ∘C, i.e., ∼0.7 GPa higher than its position calculated using the Holland and Powell (2011) data (Fig. 7). This discrepancy is equivalent to a difference in the ΔH of Reaction (R5) of 5.5 kJ, which is approximately equal to the 2σ uncertainty in the ΔH of CaSi2O5 as given by Holland and Powell (2011) (2.8 kJ). The uncertainties in ΔH for SiO2 and CaSiO3 are much smaller (0.5 kJ). Therefore, it can be concluded that the data of Holland and Powell (2011) are consistent within their 2σ uncertainty with our experimental observations. On the other hand, our results do significantly extend the pressure range of possible breyite crystallization in SiO2-saturated bulk compositions (Fig. 7).
Under natural conditions, equilibrium (Reaction R5) can be affected by the presence of TiO2. The titanite-structured CaSi2O5 phase from inclusions in diamond is enriched in TiO2 relative to coexisting breyite. This can be exemplified by the TiO2 contents of CaSiO3 (0.02 wt %) and CaSi2O5 (0.15 wt %) inclusions in diamond reported by Joswig et al. (1999). There is no direct experimental evidence on the effect of Ti on Reaction (R5). The experiments of Kubo et al. (1997) on the CaSiO3–CaTiO3 system indicate that the pressure of breyite transformation to larnite + CaSi2O5 is reduced by ∼0.7 GPa at 1200 ∘C and a CaTiSiO5 mole fraction in titanite-structured Ca(Ti,Si)SiO5 of ∼0.15. If the effect of TiO2 on Reaction (R5) is of the same order, the presence of ∼0.5 % of CaTiSiO5 in the titanite-structured phase will have a negligible effect on the breyite stability field.
5.3 Breyite crystallization under hydrous conditions
In the hydrous experiments, the iron–brucite starting mixture served as a source of fluid and an oxygen buffer. Interaction of brucite with iron produced a ferropericlase solid solution along with a reduced H2O–H2 fluid. The latter reacted with the adjacent CaCO3–SiO2 mixture to form C–O–H fluid species. The high chemical potential of H2O acted to significantly depress the melting temperature of the CaCO3–SiO2 mixture, resulting in complete melting already at 1100 ∘C and 6 GPa and very high degrees of melting at 1100 ∘C and 8–10 GPa. No separate fluid phase was observed in these experiments, indicating that all fluid components were dissolved in the melt. The hydrous melt is strongly enriched in SiO2 compared with the carbonate-dominated melt from the anhydrous experiments (Fig. 8).
The hydrous melt contained abundant suspended graphite crystals. Their nature is uncertain, and some could have formed during quenching. However, most graphite grains probably crystallized in equilibrium with the melt, which is indicated by their nonrandom distribution and very high content in some areas. The identification of equilibrium CaSi2O5 and SiO2 crystals is unambiguous because these phases always occur as euhedral crystals up to 20 µm in size, usually in the lower part of the samples (Fig. 6). In contrast, a quench nature of some breyite crystals might be suspected because of their needle-like morphology (Fig. 6). However, there are several arguments supporting a subliquidus equilibrium origin of most breyite crystals. (1) Their amount is always low and the grains are observed locally near the walls or bottom of a capsule. Here, breyite content might be locally very high (>50 vol %), whereas isolated CaSiO3 crystals were never found in the free melt volume. (2) There is a continuous transition from acicular to euhedral isometric breyite grains. The latter can hardly be interpreted as a quench phase (Fig. 6). (3) In one sample, a large isometric breyite crystal was observed between the Pt capsule and a continuous coesite layer. In this case, the local melt fraction was obviously too low to produce a large quench crystal.
Thus, our experiments demonstrated that simultaneous crystallization of breyite and diamond is possible as a result of interaction between CaCO3 and SiO2 in a metasedimentary source in the presence of reduced H2O-rich fluid or melt. Although our experiments were made in a strongly simplified system compared with real metasediments, they are directly applicable to interactions in natural environments. Both aragonite and coesite/stishovite are practically pure phases in metasediments under high-pressure, high-temperature conditions (e.g., Brey et al., 2015). Natural breyite contains up to 0.2 wt % FeO and much lower amounts of other components (Joswig et al., 1999; Burnham et al., 2015). Hence, Reactions (R2) and (R4) are not affected by the presence of other components in natural materials. Moreover, melting in natural metasediments occurs at lower temperatures compared with the experimental CaO–SiO2–CO2 system because of the fluxing influence of alkalis, which must enhance the metastability of decarbonation Reaction (R2).
The pressure range of breyite inclusion formation is limited by the diamond–graphite transformation at ∼4 GPa and 1100 ∘C and Reaction (R5) at ∼8 GPa and 1100 ∘C (Fig. 7). These conditions are slightly above the solidus of hydrous carbonated sediment similar to GLOSS (global subducting sediment) of Plank and Langmuir (1998) coexisting with graphite or diamond (Brey et al., 2015). Hence, hydrous carbonate–silicate melt (or supercritical fluid) can be obtained from subducted sediment, if the temperature of the slab is not high and does not prevent hydrous silicate (lawsonite and phengite) transport to great depths. Such conditions are characteristic of cold subducted slabs (Syracuse et al., 2010).
It is evident from the above discussion that fO2 is a very important parameter for breyite and diamond crystallization. To quantitatively evaluate a possible scenario for the formation of diamond-hosted breyite inclusions, mineral reactions in sedimentary and peridotitic materials were modeled using the dataset of Holland and Powell (2011). The calculated fO2 values are expressed in logarithmic units relative to the Fe–FeO buffer (IW).
A proposed scenario of simultaneous breyite and diamond formation at 6–8 GPa and ∼1000 ∘C is presented diagrammatically in Fig. 9. It involves melt formation in subducted sediments and its subsequent interaction with peridotite. At these depths and temperatures, the mineral assemblage of an hydrous carbonated and C-bearing sediment (GLOSS) includes coesite, kyanite, Ca-rich garnet, aragonite, lawsonite, phengite, graphite and accessory phases (Brey et al., 2015). The fO2 is controlled by the reaction
and is relatively high (logfO2 ≈ IW +5.5). The mole fraction of grossular in garnet in metasediment at 8 GPa and 1000 ∘C is 0.3–0.4 (Brey et al., 2015). Accounting for garnet composition using the Berman (1990) model for Ca–Mg–Fe garnet yields even slightly higher logfO2 values of ∼IW +5.7. Melt produced from such a metasediment will have the same fO2. It will be saturated with respect to diamond but undersaturated in breyite, because such a redox potential will provide a CO2 activity that is too high for decarbonation Reaction (R4) to proceed. However, the produced melt can be subsequently reduced through interaction with mantle peridotite, which will eventually result in diamond formation and redox freezing (Rohrbach and Schmidt, 2011). The redox conditions of mantle peridotite can be constrained by the appearance of Fe metal from below and magnesite from above, i.e., the reactions
This yields a wide range of logfO2 values, from IW to IW +4, which is more reduced than the initial metasediment-derived melt. Note that all the fO2 values imposed by Reactions (R15)–(R17) are much lower than the CCO buffer at these pressures and temperatures (CCO ≈ IW +8 at 1000–1100 ∘C and 6 GPa; Jacobsson and Oskarsson, 1994). This implies that a CO2-rich gas phase is absent, and diamond will remain stable in both metasediment and peridotite. Interaction of metasediment-derived melt with peridotite will result in reduction accompanied by Fe and Mg transfer from peridotite to melt and SiO2 transfer from melt to peridotite (Bulatov et al., 2014). If the melt is saturated in diamond (melt is derived in a diamond-bearing metasediment), its reduction will immediately result in diamond crystallization. The reduction-induced crystallization of silicate phases will begin later via two possible paths. If transport of O2 is much faster than that of Fe and Mg, melt reduction and diamond crystallization will eventually result in breyite (or CaSi2O5) crystallization and its possible entrapment by growing diamond crystals. Otherwise, in the case of rapid Fe and Mg transport, breyite formation will be suppressed, and clinopyroxene, garnet or merwinite (e.g., Sharygin et al., 2018) will form instead.
The obtained results allow us to speculate about the difference in the origins of breyite inclusions either being trapped at conditions within the breyite stability field or those that formed by retrogression of trapped CaSiO3 perovskite. Anzolini et al. (2016) emphasized the importance of the very large difference in molar volume between CaSiO3 perovskite and breyite. They argued that breyite inclusions without any evidence for diamond deformation and/or cracking were probably trapped initially as breyite. Moreover, the large volume change will result in an initial CaSiO3 perovskite inclusion converting to polymineralic aggregates containing larnite and CaSi2O5 (e.g., Joswig et al., 1999) rather than monomineralic breyite. Inspection of the phase diagram in Fig. 2 indicates that CaSiO3 inclusions coexisting with larnite or stishovite most probably had a perovskite precursor. In contrast, CaSi2O5 can be trapped together with breyite at a pressure of ∼8 GPa. Finally, the formation of breyite through reaction of CaCO3 and SiO2 implies a metasedimentary precursor, whereas CaSiO3 perovskite is a phase stable in a peridotitic mantle. Therefore, the two types of breyite inclusions should have specific geochemical and isotopic signatures. It is interesting in this respect that at least some calcium perovskite inclusions in diamond exhibit geochemical signatures of subducted oceanic crust rather than primitive or depleted peridotites (e.g., Walter et al., 2008).
All data derived from this research are presented in the enclosed Tables and Figures.
All authors contributed to the problem formation, experimental strategy and results interpretation. VKB and ABW conducted the experiments. The analytical work was performed by AVG and HEH. The manuscript was prepared by AVG and ABW.
The authors declare that they have no conflict of interest.
We are grateful to Thomas Kautz for his help in multianvil experiments and two anonymous reviewers for helpful comments and suggestions to improve the paper.
This research has been supported by the Deutsche Forschungsgemeinschaft (grant no. Wo 652/31-1) and the Russian Academy of Sciences (grant nos. 0136-2018-0028 and 0137-2019-0014).
This paper was edited by Stefano Poli and reviewed by two anonymous referees.
Akaogi, M., Yano, M., Tejima, Y., Iijima, M., and Kojitani, H.: High-pressure transitions of diopside and wollastonite: phase equilibria and thermochemistry of CaMgSi2O6, CaSiO3 and CaSi2O5–CaTiSiO5 system, Phys. Earth Planet. In., 143–144, 145–156, 2004.
Anzolini, C., Angel, R. J., Merlini, M., Derzsi, M., Tokár, K., Milani, S., Krebs, M. Y., Brenker, F. E., Nestola, F., and Harris, J. W.: Depth of formation of CaSiO3-walstromite included in super-deep diamonds, Lithos, 265, 138–147, 2016.
Anzolini, C., Prencipe, M., Alvaro, M., Romano, C., Vona, A., Lorenzon, S., Smith, E. M., Brenker, F. E., and Nestola, F.: Depth of formation of super-deep diamonds: Raman barometry of CaSiO3-walstromite inclusions, Am. Mineral., 103, 69–74, 2018.
Berman, R. G.: Mixing properties of Ca–Mg–Fe–Mn garnets, Am. Mineral., 75, 328–344, 1990.
Brenker, F., Nestola, F., Brenker, L., Peruzzo, L., Secco, L., and Harris, J. W.: Breyite, IMA 2018-062, CNMNC Newsletter No. 45, October 2018, Mineral. Mag., 82, 1225–1232, 2018.
Brenker, F. E., Vollmer, C., Vincze, L., Vekemans, B., Szymanski, A., Janssens, K., Szaloki, I., Nasdala, L., Joswig, W., and Kaminsky, F.: Carbonates from the lower part of transition zone or even the lower mantle, Earth Planet. Sc. Lett., 260, 1–9, 2007.
Brey, G. P., Bulatov, V. K., Girnis, A. V., and Lahaye, Y.: Experimental melting of carbonated peridotite at 6–10 GPa, J. Petrol., 49, 797–821, 2008.
Brey, G. P., Girnis, A. V., Bulatov, V. K., Hofer, H. E., Gerdes, A., and Woodland, A. B.: Reduced sediment melting at 7.5–12 GPa: phase relations, geochemical signals and diamond nucleation, Contrib. Mineral. Petr., 170, 18, https://doi.org/10.1007/s00410-015-1166-z, 2015
Bulanova, G. P., Walter, M. J., Smith, C. B., Kohn, S. C., Armstrong, L. S., Blundy, J., and Gobbo, L.: Mineral inclusions in sublithospheric diamonds from Collier 4 kimberlite pipe, Juina, Brazil: subducted protoliths, carbonated melts and primary kimberlite magmatism, Contrib. Mineral. Petr., 160, 489–510, 2010.
Bulatov, V. K., Brey, G. P., Girnis, A. V., Gerdes, A., and Höfer, H. E.: Carbonated sediment–peridotite interaction and melting at 7.5–12 GPa, Lithos, 200–201, 368–385, 2014.
Burnham, A. D., Thomson, A. R., Bulanova, G. P., Kohn, S. C., Smith, C. B., and Walter, M. J.: Stable isotope evidence for crustal recycling as recorded by superdeep diamonds, Earth Planet. Sc. Lett., 432, 374–380, 2015.
Burnham, A. D., Bulanova, G. P., Smith, C. B., Whitehead, S. C., Kohn, S. C., Gobbo, L., and Walter, M. J.: Diamonds from the Machado River alluvial deposit, Rondônia, Brazil, derived from both lithospheric and sublithospheric mantle, Lithos, 265, 199–213, 2016.
Carter, L. B. and Dasgupta, R.: Effect of melt composition on crustal carbonate assimilation: implications for the transition from calcite consumption to skarnification and associated CO2 degassing, Geochem. Geophy. Geosy., 17, 3893–3916, https://doi.org/10.1002/2015GC006060, 2016.
Cayzer, N. J., Odake, S., Harte, B., and Kagi, H.: Plastic deformation of lower mantle diamonds by inclusion phase transformations, Eur. J. Mineral., 20, 333–339, 2008.
Chatterjee, N. D., Johannes, W., and Leistner, H.: The system CaO–Al2O3–SiO2–H2O: new phase equilibria data, some calculated phase relations, and their petrological applications, Contrib. Mineral. Petr., 88, 1–13, 1984.
Chopin, C., Beyssac, O., Bernard, S., and Malavieille, J.: Aragonite–grossular intergrowths in eclogite-facies marble, Alpine Corsica, Eur. J. Mineral., 20, 857–865, 2008.
Davies, R. M., Griffin, W. L., O'Reilly, S. Y., and Doyle, B. J.: Mineral inclusions and geochemical characteristics of microdiamonds from the DO27, A154, A21, A418, DO18, DD17 and Ranch Lake kimberlites at Lac de Gras, Slave Craton, Canada, Lithos, 77, 39–55, 2004.
Dörsam, G., Liebscher, A., Wunder, B., Franz, G., and Gottschalk, M.: Crystal structure refinement of synthetic Ca0.43Sr0.57[SiO3]-walstromite and walstromite–fluid Ca–Sr distribution at upper-mantle conditions, Eur. J. Mineral., 21, 705–714, 2009.
Essene, E.: High-pressure transformations in CaSiO3, Contrib. Mineral. Petr., 45, 247–250, 1974.
Fedoraeva, A. S., Shatskiy, A., and Litasov, K. D.: The join CaCO3–CaSiO3 at 6 GPa with implication to Ca-rich lithologies trapped by kimberlitic diamonds, High Pressure Res., 39, https://doi.org/10.1080/08957959.2019.1660325, 2019.
Galvez, M. E., Beyssac, O., Martinez, I., Benzerara, K., Chaduteau, C., Malvoisin, B., and Malavieille, J.: Graphite formation by carbonate reduction during subduction, Nat. Geosci., 6, 473–477, 2013a.
Galvez, M. E., Martinez, I., Beyssac, O., Benzerara, K., Agrinier, P., and Assayag, N.: Metasomatism and graphite formation at a lithological interface in Malaspina (Alpine Corsica, France), Contrib. Mineral. Petr., 166, 1687–1708, 2013b.
Gasparik, T., Wolf, K., and Smith, C. M.: Experimental determination of phase relations in the CaSiO3 system from 8 to 15 GPa, Am. Mineral., 79, 1219–1222, 1994.
Grassi, D. and Schmidt, M. W.: Melting of carbonated pelites at 8–13 GPa: generating K-rich carbonatites for mantle metasomatism, Contrib. Mineral. Petr., 162, 169–191, 2011a.
Grassi, D. and Schmidt, M. W.: The melting of carbonated pelites from 70 to 700 km depth, J. Petrol., 52, 765–789, 2011b.
Hammouda, T.: High-pressure melting of carbonated eclogite and experimental constraints on carbon recycling and storage in the mantle, Earth Planet. Sc. Lett., 214, 357–368, 2003.
Harte, B.: Diamond formation in the deep mantle: the record of mineral inclusions and their distribution in relation to mantle dehydration zones, Mineral. Mag., 74, 189–215, 2010.
Hasterok, D. and Chapman, D. S.: Heat production and geotherms for the continental lithosphere, Earth Planet. Sc. Lett., 307, 59–70, 2011.
Hayman, P. C., Kopylova, M. G., and Kaminsky, F. V.: Lower mantle diamonds from Rio Soriso (Juina area, Mato Grosso, Brazil), Contrib. Mineral. Petr., 149, 430–445, 2005.
Holland, T. J. B. and Powell, R.: An improved and extended internally consistent thermodynamic dataset for phases of petrological interest, involving a new equation of state for solids, J. Metamorph. Geol., 29, 333–383, 2011.
Huang, W.-L., Wyllie, P. J., and Nehru, C. E.: Subsolidus and liquidus phase relationships in the system CaO–SiO2–CO2 to 30 kbar with geological applications, Am. Mineral., 65, 285–301, 1980.
Irifune, T. and Tsuchiya, T.: Mineralogy of the Earth – phase transitions and mineralogy of the lower mantle, Treatise on Geophysics, 2, 33–62, 2007.
Jacobsson, S. and Oskarsson, N.: The system C–O in equilibrium with graphite at high pressure and temperature: An experimental study, Geochim. Cosmochim. Ac., 58, 9–17, 1994.
Joswig, W., Stachel, T., Harris, J. W., Baur, W. H., and Brey, G. P.: New Ca-silicate inclusions in diamonds – tracers from the lower mantle, Earth Planet. Sc. Lett., 173, 1–6, 1999.
Joswig, W., Paulus, E. F., Winkler, B., and Milman, V.: The crystal structure of CaSiO3-walstromite, a special isomorph of wollastonite-II, Z. Kristallogr., 218, 811–818, 2003.
Kaminsky, F.: Mineralogy of the lower mantle: a review of “super-deep” mineral inclusions in diamond, Earth-Sci. Rev., 110, 127–147, 2012.
Kaminsky, F., Wirth, R., Matsyuk, S., Schreiber, A., and Thomas, R.: Nyerereite and nahcolite inclusions in diamond: evidence for lower-mantle carbonatitic magmas, Mineral. Mag., 73, 797–816, 2009.
Kaminsky, F., Matzel, J., Jacobsen, B., Hutcheon, I., and Wirth, R.: Isotopic fractionation of oxygen and carbon in decomposed lower-mantle inclusions in diamond, Miner. Petrol., 110, 379–385, 2016.
Kanzaki, M., Stebbins, J. F., and Xue, X.: Characterization of quenched high pressure phases in CaSiO3 system by XRD and 29Si NMR, Geophys. Res. Lett., 18(3), 463–466, 1991.
Kennedy, C. S. and Kennedy, G. C.: The equilibrium boundary between graphite and diamond, J. Geophys. Res., 81, 2467–2470, 1976.
Kiseeva, E. S., Yaxley, G. M., Hermann, J., Litasov, K. D., Rosenthal, A., and Kamenetsky, V. S.: An experimental study of carbonated eclogite at 3.5–5.5 GPa – Implications for silicate and carbonate metasomatism in the cratonic mantle, J. Petrol., 53, 727–759, 2012.
Kiseeva, E. S., Litasov, K. D., Yaxley, G. M., Ohtani, E., and Kamenetsky, V. S.: Melting and phase relations of carbonated eclogite at 9–21 GPa and the petrogenesis of alkali-rich melts in the deep mantle, J. Petrol., 54, 1555–1583, 2013.
Knoche, R., Angel, R. J., Seifert, F., and Fliervoet, T. F.: Complete substitution of Si for Ti in titanite Ca(Ti1−xSix)VISiIVO5, Am. Mineral., 83, 1168–1175, 1998.
Kubo, A., Suzuki, T., and Akaogi, M.: High pressure phase equilibria in the system CaTiO3–CaSiO3: stability of perovskite solid solutions, Phys. Chem. Miner., 24, 488–494, 1997.
Li, Y.-H. and Schoonmaker, J. E.: Chemical composition and mineralogy of marine sediments, Treatise on Geochemistry, 7, 1–35, 2003.
Li, Z., Li, J., Lange, R., Liu, J., and Militzer, B.: Determination of calcium carbonate and sodium carbonate melting curves up to Earth's transition zone pressures with implications for the deep carbon cycle, Earth Planet. Sc. Lett., 457, 395–402, 2017.
Litasov, K. D., Shatskiy, A., Gavryushkin, P. N., Bekhtenova, A. E., Dorogokupets, P. I., Danilov, B. S., Higo, Y., Akilbekov, A. T., and Inerbaev, T. M.: P–V–T equation of state of CaCO3 aragonite to 29 GPa and 1673 K: In situ X-ray diffraction study, Phys. Earth Planet. In., 265, 82–91, 2017.
Liu, J., Topor, L., Zhang, J., Navrotsky, A., and Liebermann, R.: Calorimetric study of the coesite–stishovite transformation and calculation of the phase boundary, Phys. Chem. Miner., 23, 11–16, 1996.
Mann, U. and Schmidt, M. W.: Melting of pelitic sediments at subarc depths: 1. Flux vs. fluid-absent melting and a parameterization of melt productivity, Chem. Geol., 404, 150–167, 2015.
Martin, L. A. J. and Hermann, J.: Experimental Phase relations in altered oceanic crust: implications for carbon recycling at subduction zones, J. Petrol., 59, 299–320, 2018.
Morishima, H., Kato, T., Suto, M., Ohtani, E., Urakawa, S., Utsumi, W., Shimomura, O., and Kikegawa, T.: The phase boundary between α- and ß-Mg2SiO4 determined by in situ X-ray observation, Science, 265, 1202–1203, 1994.
Nasdala, L., Brenker, F. E., Glinnemann, J., Hofmeister, W., Gasparik, T., Harris, J. W., Stachel, T., and Reese, I.: Spectroscopic 2D-tomography: Residual pressure and strain around mineral inclusions in diamonds, Eur. J. Mineral., 15, 931–935, 2003.
O'Neill, H. S. C., Pownceby, M. I., and McCammon, C. A.: The magnesiowustite: iron equilibrium and its implications for the activity composition relations of (Mg,Fe)2SiO4 olivine solid solutions, Contrib. Mineral. Petr., 146, 308–325, 2003.
Pearson, D. G., Brenker, F. E., Nestola, F., McNeill, J., Nasdala, L., Hutchison, M. T., Matveev, S., Mather, K., Silversmit, G., Schmitz, S., Vekemans, B., and Vincze, L.: Hydrous mantle transition zone indicated by ringwoodite included within diamond, Nature, 507, 221–224, 2014.
Perrillat, J.-P., Ricolleau, A., Daniel, I., Fiquet, G., Mezouar, M., Guignot, N., and Cardon, H.: Phase transformations of subducted basaltic crust in the upmost lower mantle, Phys. Earth Planet. In., 157, 139–149, 2006.
Plank, T. and Langmuir, C. H.: The chemical composition of subducting sediment and its consequences for the crust and mantle, Chem. Geol., 145, 325–394, 1998.
Poli, S.: Carbon mobilized at shallow depths in subduction zones by carbonatitic liquids, Nat. Geosci., 8, 633–637, 2015.
Ringwood, A. E. and Major, A.: Some high-pressure transformations of geophysical significance, Earth Planet. Sc. Lett., 2, 106–110, 1967.
Rohrbach, A. and Schmidt, M. W.: Redox freezing and melting in the Earth's deep mantle resulting from carbon–iron redox coupling, Nature, 472, 209–212, 2011.
Ross, N. L., Akaogi, M., Navrotsky, A., Susaki, J., and McMillan, P.: Phase transitions among the CaGeO3 polymorphs (wollastonite, garnet, and perovskite structures): studies by high-pressure synthesis, high-temperature calorimetry, and vibrational spectroscopy and calculation, J. Geophys. Res., 91, 4685–4696, 1986.
Sharygin, I. S., Shatskiy, A., Litasov, K. D., Golovin, A. V., Ohtani, E., and Pokhilenko, N. P.: Interaction of peridotite with Ca-rich carbonatite melt at 3.1 and 6.5 GPa: Implication for merwinite formation in upper mantle, and for the metasomatic origin of sublithospheric diamonds with Ca-rich suite of inclusions, Contrib. Mineral. Petr., 173, https://doi.org/10.1007s00410-015-1166-z, 2018.
Stachel, T., Harris, J. W., Brey, G. P., and Joswig, W.: Kankan diamonds (Guinea) II: lower mantle inclusion parageneses, Contrib. Mineral. Petr., 140, 16–27, 2000.
Sueda, Y., Irifune, T., Yamada, A., Inoue, T., Liu, X., and Funakoshi K.: The phase boundary between CaSiO3 perovskite and Ca2SiO4+CaSi2O5 determined by in situ X-ray observations, Geophys. Res. Lett., 33, L10307, https://doi.org/10.1029/2006GL025772, 2006.
Syracuse, E. M., van Keken, P. E., and Abers, G. A.: The global range of subduction zone thermal models, Phys. Earth Planet. In., 183, 73–90, 2010.
Tappert, R., Stachel, T., Harris, J. W., Shimizu, N., and Brey, G. P.: Mineral inclusions in diamonds from the Panda kimberlite, Slave Province, Canada, Eur. J. Mineral., 17, 423–440, 2005.
Tsuno, K. and Dasgupta, R.: The effect of carbonates on near-solidus melting of pelite at 3 GPa: Relative efficiency of H2O and CO2 subduction, Earth Planet. Sc. Lett., 319–320, 185–196, 2012.
Walter, M. J., Bulanova, G. P., Armstrong, L. S., Keshav, S., Blundy, J. D., Gudfinnson, G., Lord, O. T., Lennie, A. R., Clark, S. M., Smith, C. B., and Gobbo, L.: Primary carbonatite melt from deeply subducted oceanic crust, Nature, 454, 622–625, 2008.
Wirth, R., Kaminsky, F., Matsyuk, S., and Schreiber, A.: Unusual micro- and nano-inclusions in diamonds from the Juina Area, Brazil, Earth Planet. Sc. Lett., 286, 292–303, 2009.
Wood, B. J.: Phase transformations and partitioning relations in peridotite under lower mantle conditions. Earth Planet. Sc. Lett., 174, 341–354, 2000.
Woodland, A. B., Bulatov, V. K., Brey, G. P., Girnis, A. V., Höfer, H. E., and Gerdes, A.: Subduction factory in an ampoule: Experiments on sediment–peridotite interaction under temperature gradient conditions, Geochim. Cosmochim. Ac., 223, 319–349, 2018.
Wyllie, P. J. and Huang, W.-L.: Carbonation and melting reactions in the system CaO–MgO–SiO2–CO2 at mantle pressures with geophysical and petrological applications, Contrib. Mineral. Petr., 54, 79–107, 1976.
Yaxley, G. M. and Brey, G. P.: Phase relations of carbonate-bearing eclogite assemblages from 2.5 to 5.5 GPa: implications for petrogenesis of carbonatites, Contrib. Mineral. Petr., 146, 606–619, 2004.
Zedgenizov, D. A., Shatsky, V. S., Ragozin, A. L., Panin, A. V., Evtushenko, O. V., and Kagi, H.: Evidence for phase transitions in mineral inclusions in superdeep diamonds of the Sao Luiz deposit (Brazil), Rus. Geol. Geophys+., 56, 296–305, 2015.
Zhang, J., Li, B., Utsumi, W., and Liebermann, R. C.: In situ X-ray observations of the coesite–stishovite transition: reversed phase boundary and kinetics, Phys. Chem. Miner., 23, 1–10, 1996.