the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 26 Jun 2024
| 26 Jun 2024
Fluor-rewitzerite, [(H2O)K]Mn2(Al2Ti)(PO4)4(OF)(H2O)10 ⋅ 4H2O, a new paulkerrite-group mineral, from the Hagendorf-Süd pegmatite, Oberpfalz, Bavaria, Germany
Rupert Hochleitner
Ian E. Grey
Anthony R. Kampf
Stephanie Boer
Colin M. MacRae
William G. Mumme
Nicholas C. Wilson
Fluor-rewitzerite, [(H2O)K]Mn2(Al2Ti)(PO4)4(OF)(H2O)10⋅4H2O, is a new monoclinic member of the paulkerrite group, from the Hagendorf-Süd pegmatite, Oberpfalz (Upper Palatinate in English), Bavaria, Germany. It occurs on the walls of vugs in corroded zwieselite, in association with Zn- and Al-bearing earlshannonite, fluorapatite, jahnsite-(CaMnMn) and Al-rich strunzite. Fluor-rewitzerite forms clusters of colourless stubby prisms up to 0.1 mm long that are flattened on {010}; elongated along [100]; and show the forms {100}, {010}, {001}, {111} and . Twinning occurs by 2-fold rotation about c. The measured density is 2.42(2) g cm−3. Optically, fluor-rewitzerite crystals are biaxial (+), with α = 1.569(3), β = 1.582(3), γ = 1.602(3) (white light) and 2V(meas) = 78(1)°. The empirical formula from electron microprobe analyses and structure refinement is A1[(H2O)0.85K0.15]Σ1.00A2(K1.00) M1(Mn2+1.50Mg0.09Fe2+0.41)Σ2.00M2+M3(Al1.70Ti4+0.89Fe3+0.42)Σ3.01(PO4)3.99X(O1.09F0.92)Σ2.01(H2O)10⋅4.12H2O. Fluor-rewitzerite has monoclinic symmetry with space group P21/c and unit-cell parameters a = 10.407(1) Å, b = 20.514(2) Å, c = 12.193(1) Å, β = 90.49(2)°, V = 2603.0(4) Å3 and Z = 4. The crystal structure was refined using synchrotron single-crystal data to Robs=0.058 for 6186 reflections with I>3σ(I). Fluor-rewitzerite is the fluoride analogue of rewitzerite, with F dominant over OH at the X sites of the general formula A1A2M12M22M3(PO4)4X2(H2O)10⋅4H2O.
- Article
(3500 KB) - Full-text XML
-
Supplement
(680 KB) - BibTeX
- EndNote





Rewitzerite, [K(H2O)]Mn2(Al2Ti)(PO4)4[O(OH)](H2O)10⋅4H2O, was recently approved as a new monoclinic mineral of the paulkerrite group (Grey et al., 2023a). The ideal formula for rewitzerite conforms to the general formula for the monoclinic group members, A1A2M12M22M3(PO4)4X2(H2O)10⋅4H2O, where A = K, H2O, □ (denoting a vacancy); M1 = Mn2+, Mg, Fe2+, Zn (rarely Fe3+); M2 and M3 = Fe3+, Al, Ti4+; and X = O, OH, F. A high degree of mixing of Fe3+, Al and Ti4+ occurs at the M2 and M3 sites, and charge balance is maintained for different ratios at M2 and M3 by variations in the ratios at the X sites. For end-member rewitzerite, with M22M3 = Al2Ti, co-dominant OH− and O2− are at the X sites.
In ongoing studies of potential new secondary phosphate minerals in the Erich Keck collection at the Bavarian State Collection for Mineralogy, Munich, using scanning electron microscopy coupled with energy-dispersive X-ray analysis, we identified a specimen as having an analysis very similar to that for rewitzerite but with more than twice the F content. More detailed studies, including electron microprobe analyses and single-crystal X-ray diffraction, confirmed the mineral as the fluoride analogue of rewitzerite with F dominant over OH at the X sites in the crystal structure. The mineral and its name have been approved by the International Mineralogical Association (IMA) Commission on New Minerals, Nomenclature and Classification (CNMNC), proposal IMA-2023-115.
The holotype specimen is housed in the mineralogical collections of the Bavarian State Mineral Collection, Munich, registration number MSM 80824. A cotype specimen used for the optical properties, powder X-ray diffraction (XRD) and Raman spectrum is located at the Natural History Museum of Los Angeles County, catalogue number 76307.
Fluor-rewitzerite was found in a recent study of specimens of altered zwieselite that were collected by Erich Keck in the mid-1970s from the 67 m level of the Hagendorf-Süd pegmatite mine quarry in the Oberpfalz (Upper Palatinate in English), northeast Bavaria (49°39′1′′ N, 12°27′35′′ E). The holotype specimen consists of altered light-brown zwieselite with many vugs up to 5 mm in diameter. The vugs are lined with tiny (<0.1 mm) colourless crystals of fluor-rewitzerite sitting directly on the corroded zwieselite. Associated minerals are fluorapatite, white needles of Al-rich strunzite, tabular yellow crystals of Na-bearing jahnsite-(CaMnMn) and yellow microspheres of Zn- and Al-bearing earlshannonite. A massive black mineral filling some vugs has been identified by powder XRD as a Zn-bearing cryptomelane. Directly sitting on the crystals of fluor-rewitzerite are tiny white balls of iangreyite as the youngest member of the association. Directly embedded in the zwieselite and sometimes protruding into the corrosion vugs – and thus older than the fluor-rewitzerite – are massive sphalerite, tabular crystals of columbite and small grains of pyrite.
Fluor-rewitzerite forms clusters of colourless stubby prisms up to 0.1 mm long (Figs. 1 and 2). The crystals are flattened on {010}; elongated along [100]; and show the forms {100}, {010}, {001}, {111} and (Fig. 2). Twinning occurs by 2-fold rotation about c. The density, measured by the flotation method in mixtures of methylene iodide and toluene, is 2.42(2) g cm−3, compared with a value of 2.418 g cm−3 calculated for the empirical formula and single-crystal unit-cell parameters.

Figure 1Fluor-rewitzerite crystals in a vug in altered zwieselite. The field of view (FOV) is 0.60 mm. Photo by Christian Rewitzer.
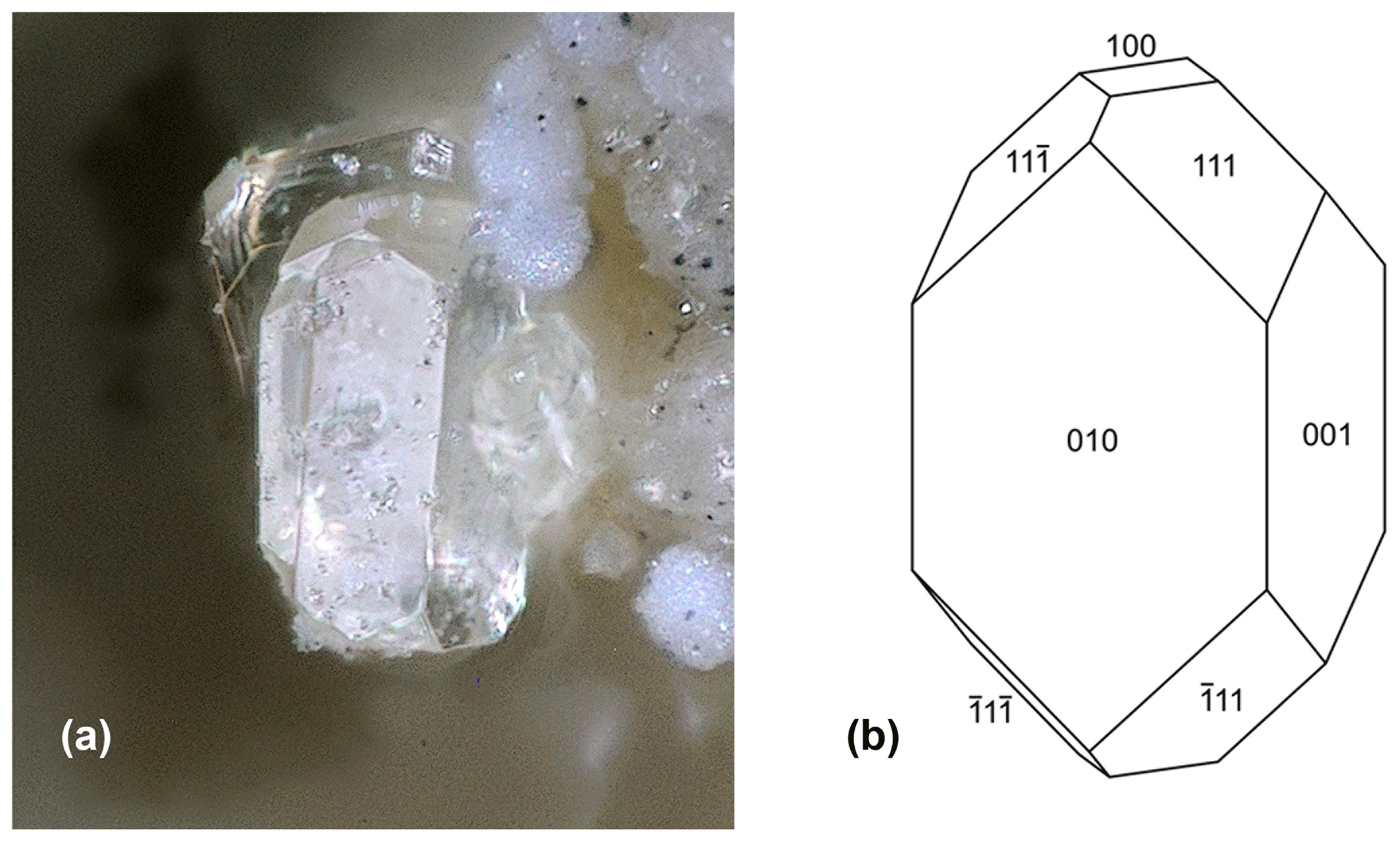
Figure 2(a) Fluor-rewitzerite crystal, FOV 0.10 mm (photo Christian Rewitzer). (b) Crystal drawing of fluor-rewitzerite crystal.
Optically, fluor-rewitzerite crystals are biaxial (+), with α = 1.569(3), β = 1.582(3) and γ = 1.6021(3) (measured in white light). The measured 2V from extinction data analysed with EXCALIBR (Gunter et al., 2004) is 78(1)°, and the calculated 2V is 78.6°. Dispersion was not observed. The optical orientation is X = b, Y = c and Z = a. The Gladstone–Dale compatibility index (Mandarino, 1981) is 0.035 (excellent) based on the empirical formula and the measured density.
Crystals of fluor-rewitzerite were analysed using wavelength-dispersive electron microprobe (EMP) spectrometry on a JEOL JXA-8530F Hyperprobe operated at an accelerating voltage of 15 kV and a beam current of 2.0 nA. The beam was defocused to between 8 and 10 µm. Both specimen and standards were coated with a 25 Å thick film of iridium for the analyses. Highly hydrated paulkerrite-group minerals present problems for analysis because of dehydration in the high vacuum of the microprobe, resulting in severe cracking and higher analytical totals (Sejkora et al., 2006). To minimise this effect, a cold stage cooled to liquid nitrogen temperature was employed in the microprobe and the specimen was precooled under dry nitrogen prior to introduction to the microprobe vacuum.
Analytical results (average of 13 analyses on four crystals) are given in Table 1. There was insufficient material for direct determination of H2O, so it was based upon the crystal structure and stoichiometry (15 H2O per 4 P). The calculated water content was iterated in the matrix correction procedure. The EMP results show a negative correlation between Al2O3 and Fe2O3 (R2 = 0.66) and a weak positive correlation between Al2O3 and F (R2 = 0.40). No significant correlations were found for K and Ti with other elements. The analysis results for rewitzerite (Grey et al., 2023a) are included in Table 1 for comparison. Fluor-rewitzerite has 50 % more K2O than, 80 % more MnO than and more than twice the F content of rewitzerite but has lower TiO2 and MgO contents.
Table 1Analytical data (wt %) for fluor-rewitzerite.
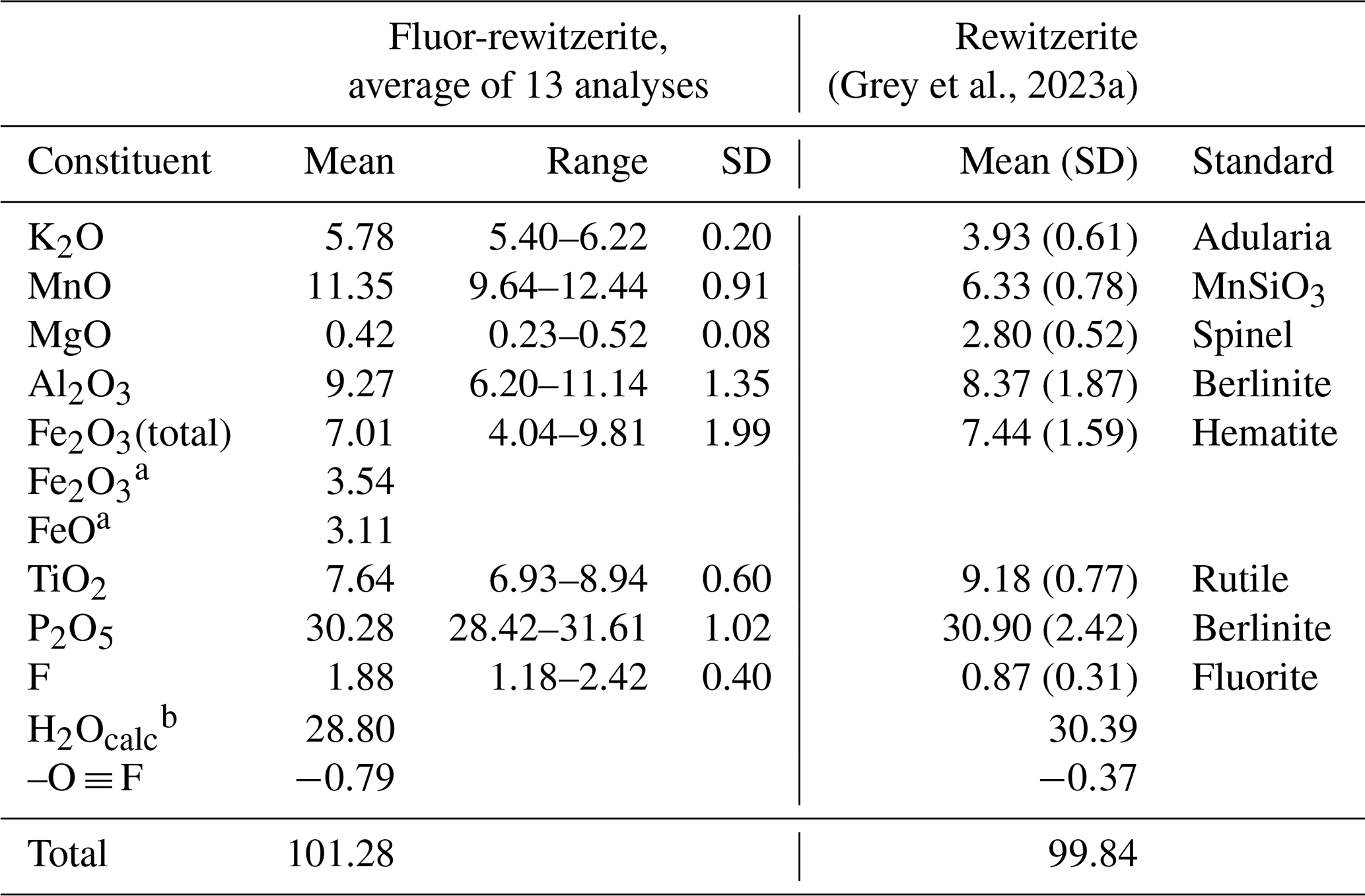
a Based on Fe2+ at M1 and Fe3+ at the M2 and M3 sites. b Based on stoichiometry: 15 H2O per 4 P.
The atoms per formula unit (apfu), normalised to 9() apfu, are as follows:
Expressing the apfu in structural form (general formula given in the Introduction) and grouping the M2 and M3 sites using the merged site procedure described by Grey et al. (2023b) gives the following empirical formula:
The simplified formula is
The ideal formula is [(H2O)K]Mn2(Al2Ti)(PO4)4(OF)(H2O)10⋅4H2O, which requires K2O 5.03, MnO 15.16, Al2O3 10.90, P2O5 30.34, TiO2 8.54, F 2.03, H2O 28.85, –O ≡ F −0.85 and total 100.00 wt %.
Note that the merged (M22M3) sites approach is illustrated graphically in Fig. 4, showing a ternary Al3–Fe–Ti3 diagram with the possible end-member compositions designated (Ti3, Al2Ti, Ti2Al, etc.) and the empirical compositions for published paulkerrite-group minerals shown by crosses. The empirical (M22M3) composition for fluor-rewitzerite is shown in Fig. 4 to be located in the end-member (Al2Ti) composition field. Combining this with the dominant species at the A, M1 and X sites leads to the end-member (ideal) formula reported above.
Raman spectroscopy was conducted on a Horiba XploRA PLUS spectrometer using a 532 nm diode laser, 100 µm slit, 1800 grooves mm−1 diffraction grating and a 100× (0.9 numerical aperture) objective. The spectrum, shown in Fig. 3, is almost identical to that for rewitzerite and the same assignments apply as given for rewitzerite (Grey et al., 2023a). The O–H stretch region has a broad peak that can be assigned to H-bonded water, with a maximum at 3355 cm−1 and a shoulder at 3115 cm−1. Hydroxyl ion stretching is evident by a shoulder at 3570 cm−1. The H–O–H bending mode region for water has a peak at 1650 cm−1. Three strong peaks at 955, 970 and 1015 cm−1 in the P–O stretching region can be assigned to symmetric stretching modes, while weaker peaks at 1095 and 1130 cm−1 correspond to antisymmetric P–O stretching modes. Bending mode vibrations of the (PO4)3− groups are located at 600 cm−1 and at 425, 445 and 480 cm−1. Peaks at lower wavenumbers are related to lattice vibrations. A strong peak at 835 cm−1 with a shoulder at 780 cm−1 can be assigned to Ti–O stretch vibrations for short Ti–O bonds that occur in linear trimers of corner-connected octahedra M2–M3–M2 in the structure, by analogy with published Raman spectra for titanates containing short Ti–O distances (Tu et al., 1996; Bamberger et al., 1990; Silva et al., 2018).
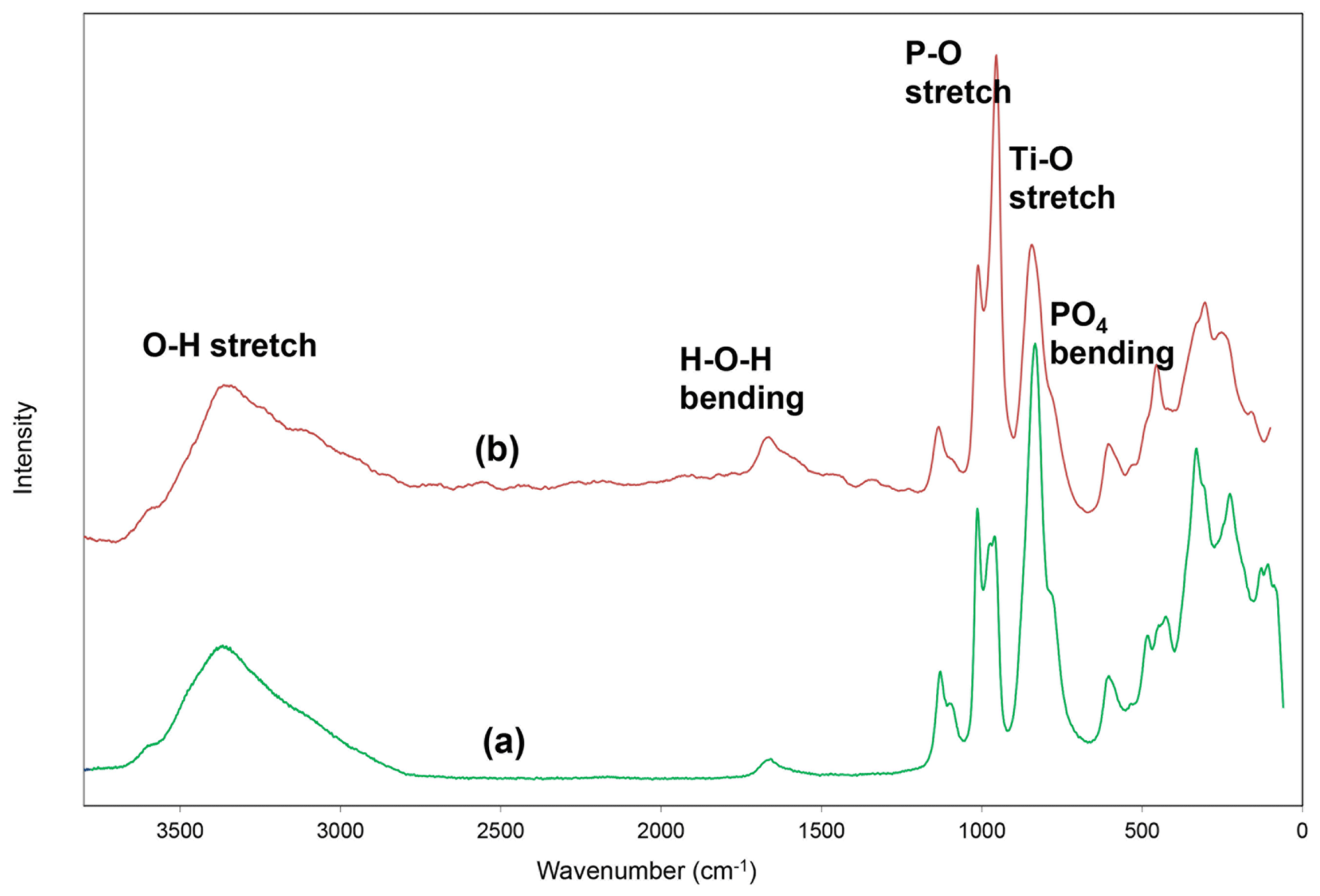
Figure 3Raman spectrum of (a) fluor-rewitzerite and (b) rewitzerite (Grey et al., 2023a).
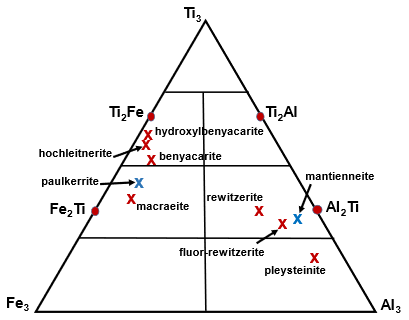
Figure 4Ternary diagram for (M2)2M3 site Al–Ti–Fe3+ compositions, showing end-member compositions (Al2Ti, Ti2Al, etc.) and location of the empirical composition for fluor-rewitzerite, For comparison the published empirical compositions are shown for the paulkerrite-group minerals benyacarite (Demartin et al., 1997), hochleitnerite (Grey et al., 2023d), hydroxylbenyacarite (Hochleitner et al., 2024), mantienneite (Fransolet et al., 1984), paulkerrite (Peacor et al., 1984), pleysteinite (Grey et al., 2023c), rewitzerite (Grey et al., 2023a) and macraeite (Grey et al., 2024). Red crosses correspond to minerals with Mn at M1, and blue crosses correspond to minerals with Mg at M1.
6.1 X-ray powder diffraction
X-ray powder diffraction data were recorded using a Rigaku R-Axis Rapid II curved-imaging-plate microdiffractometer with monochromatised MoKα radiation. A Gandolfi-like motion on the ϕ and ω axes was used to randomise the sample. Observed d values and intensities were derived by profile fitting using JADE Pro software (Materials Data, Inc.). Data are given in Table 2. Refined monoclinic unit-cell parameters (space group P2(no. 14)) are a = 10.425(18) Å, b = 20.591(18) Å, c = 12.285(18) Å, β = 90.47(10)°, V = 2637(6) Å3 and Z = 4.

6.2 Synchrotron single-crystal diffraction
A crystal measuring 0.040 × 0.050 × 0.070 mm3 was used for a single-crystal diffraction data collection at the Australian Synchrotron microfocus beamline MX2 (Aragao et al., 2018). Intensity data were collected using a Dectris Eiger 16M detector and monochromatic radiation with a wavelength of 0.7109 Å. The crystal was maintained at 100 K in an open-flow nitrogen cryostream during data collections. The diffraction data were collected using a single 36 s sweep of 360° rotation around ϕ. The resulting dataset consists of 3600 individual images with an approximate ϕ angle of each image being 0.1°. The raw intensity dataset was processed using XDS software to produce data files that were analysed using SHELXT (Sheldrick, 2015) and JANA2006 (Petříček et al., 2014). Refined unit-cell parameters and other data collection details are given in Table 3.
Table 3Crystal data and structure refinement for fluor-rewitzerite.

A structural model for fluor-rewitzerite was obtained in space group P2 using SHELXT (Sheldrick, 2015). The model had the same structure as for rewitzerite (Grey et al., 2023a), and so the rewitzerite coordinate file was used to initiate the refinement to ensure the same atom labelling. Twinning was implemented with 2-fold rotation about c. Consistent with the model for rewitzerite, Mn plus Mg was incorporated at the M1 sites, Al and Ti at the M2 and M3 sites, and K and O (for H2O) at the A sites, and their occupancies were refined to obtain the site scattering. Refinement of the ratio at the A sites gave K occupancies lower than the K content analysed by electron microprobe (EMP). A likely explanation for this discrepancy is that the A sites also contain vacancies, as reported for other paulkerrite-group minerals (Hochleitner et al., 2024; Rewitzer et al., 2024). Adjusting the vacancy content gave a reasonable match to the EMP-derived K content for 0.1 vacancies per A site.
Difference Fourier maps were used to locate H atoms. A total of 25 H atoms were unambiguously located of a possible 30 H atoms from 15 independent H2O groups. The H atoms were refined with soft restraints (O–H = 0.85(1) Å and H–O–H = 109.47(5)°).
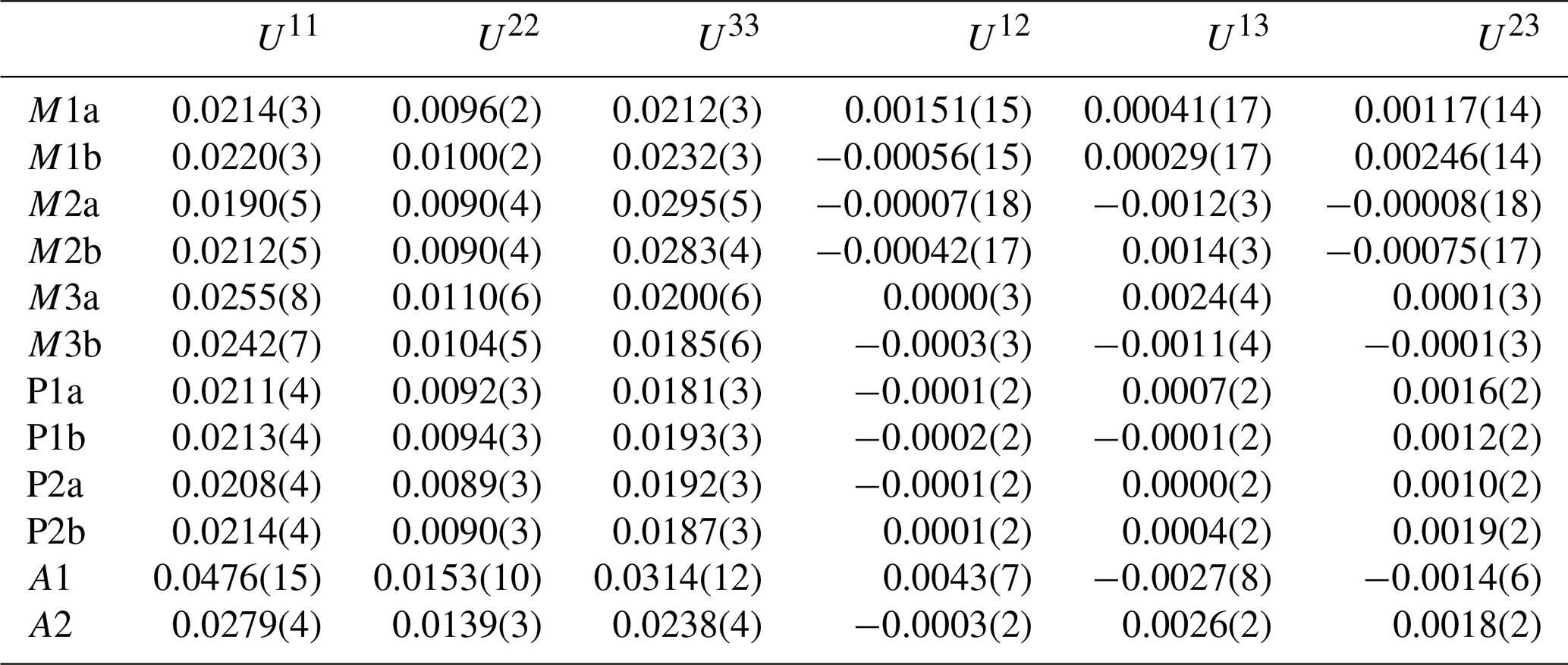
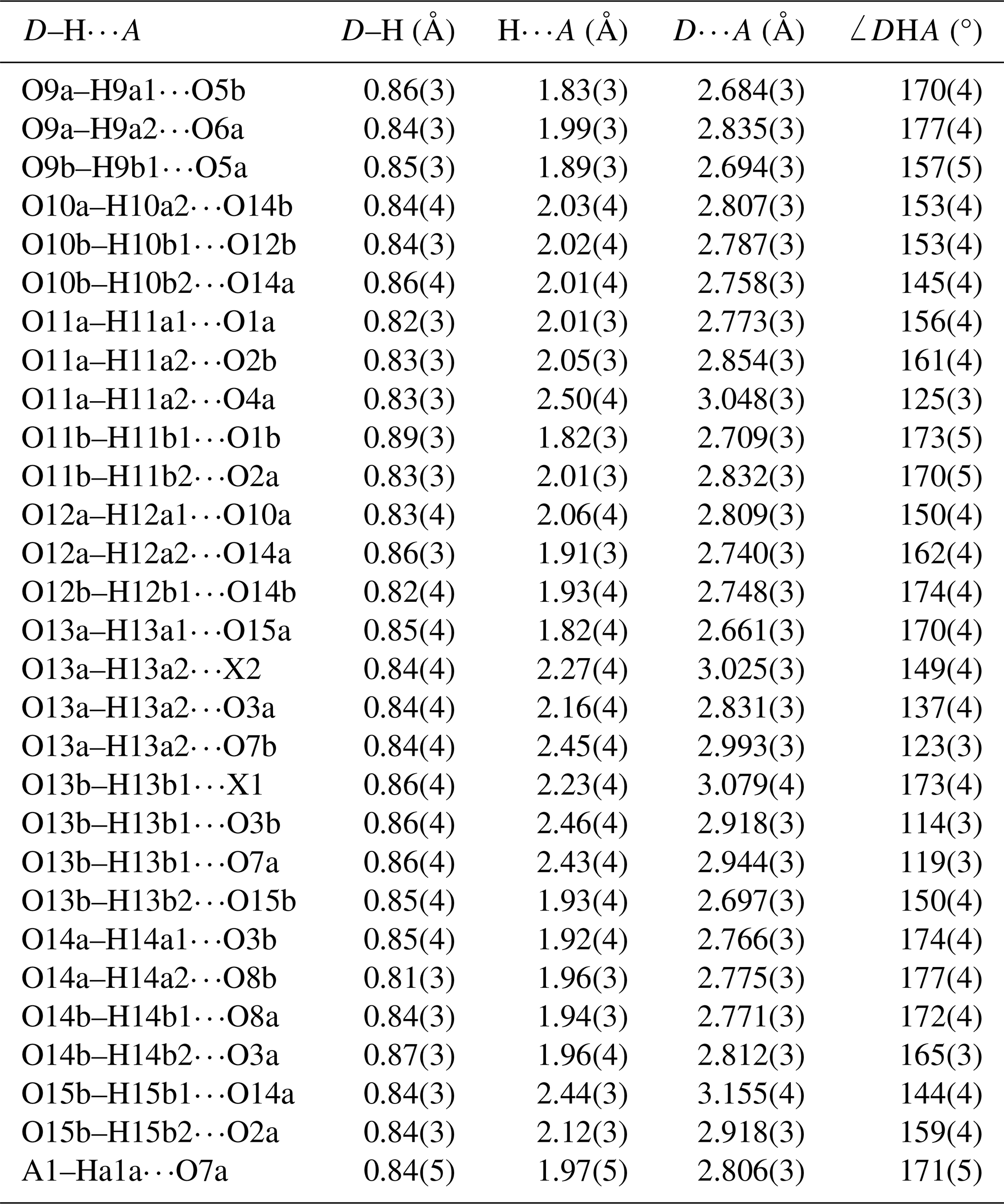
Refinement with anisotropic displacement parameters for all non-H atoms and an overall isotropic parameter for Hs in JANA2006 converged at Robs=0.058 for 6186 reflections with I>3σI. Details of the data collection and refinement are given in Table 3. The refined coordinates, equivalent isotropic displacement parameters and bond valence sum (BVS) values (Gagné and Hawthorne, 2015) from the refinement are reported in Table 4. Refined site scattering values are in Table 5. Anisotropic displacement parameters for the metal atoms are given in Table 6, and H bonds are reported in Table 7. Selected interatomic distances are reported in Table 8.
Table 4Refined atom coordinates, equivalent isotropic displacement parameters (Å2) and bond valence sums (BVSs, in valence units) for fluor-rewitzerite.
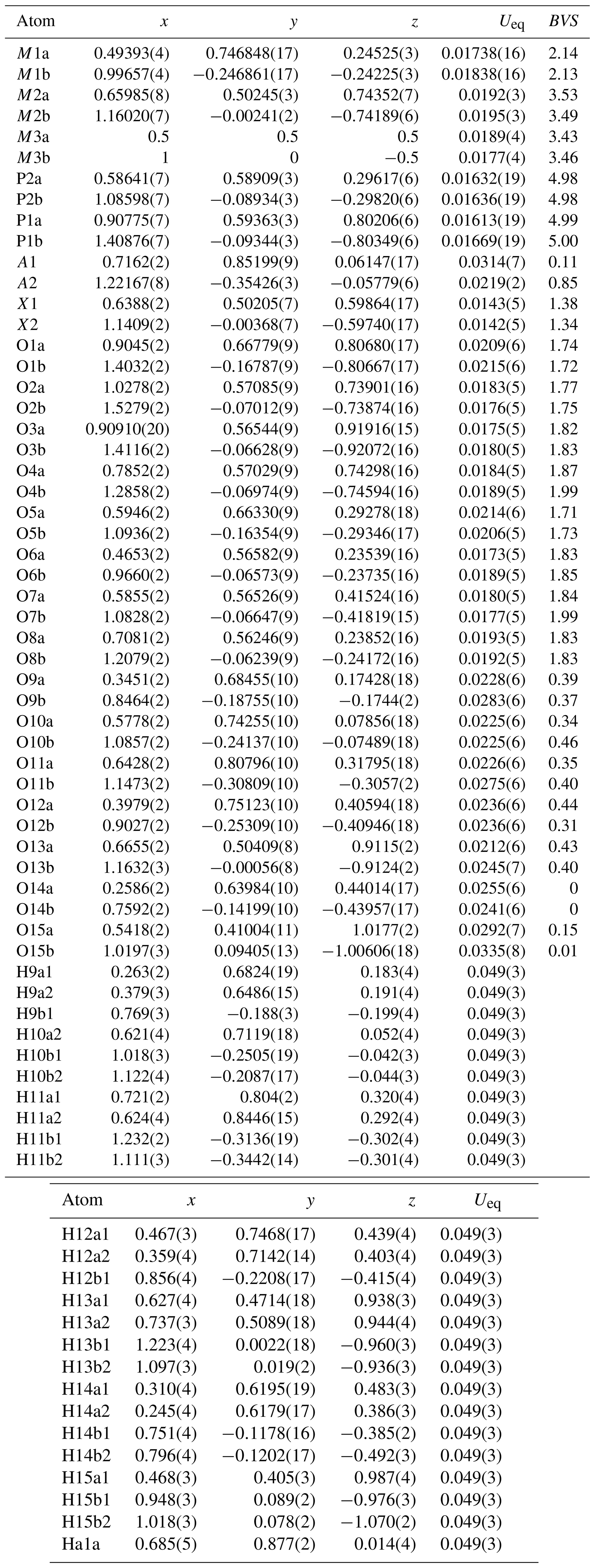
Table 5Refined site scattering for fluor-rewitzerite.
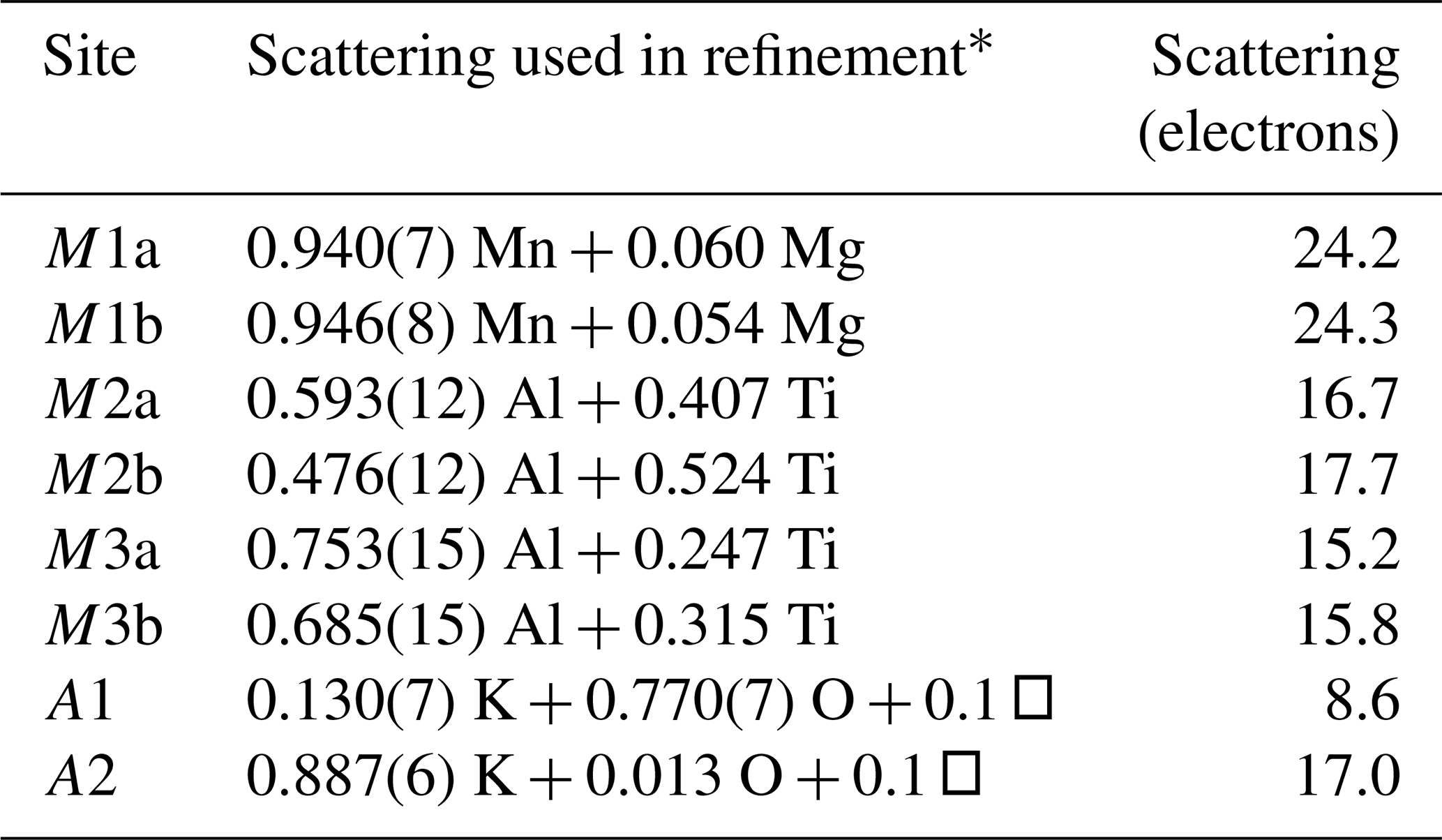
* Mn scattering curve used for Mn plus minor Fe; Ti scattering curve used for Ti plus minor Fe.
Table 6Anisotropic displacement parameters (Å2) for metal atom sites in fluor-rewitzerite.
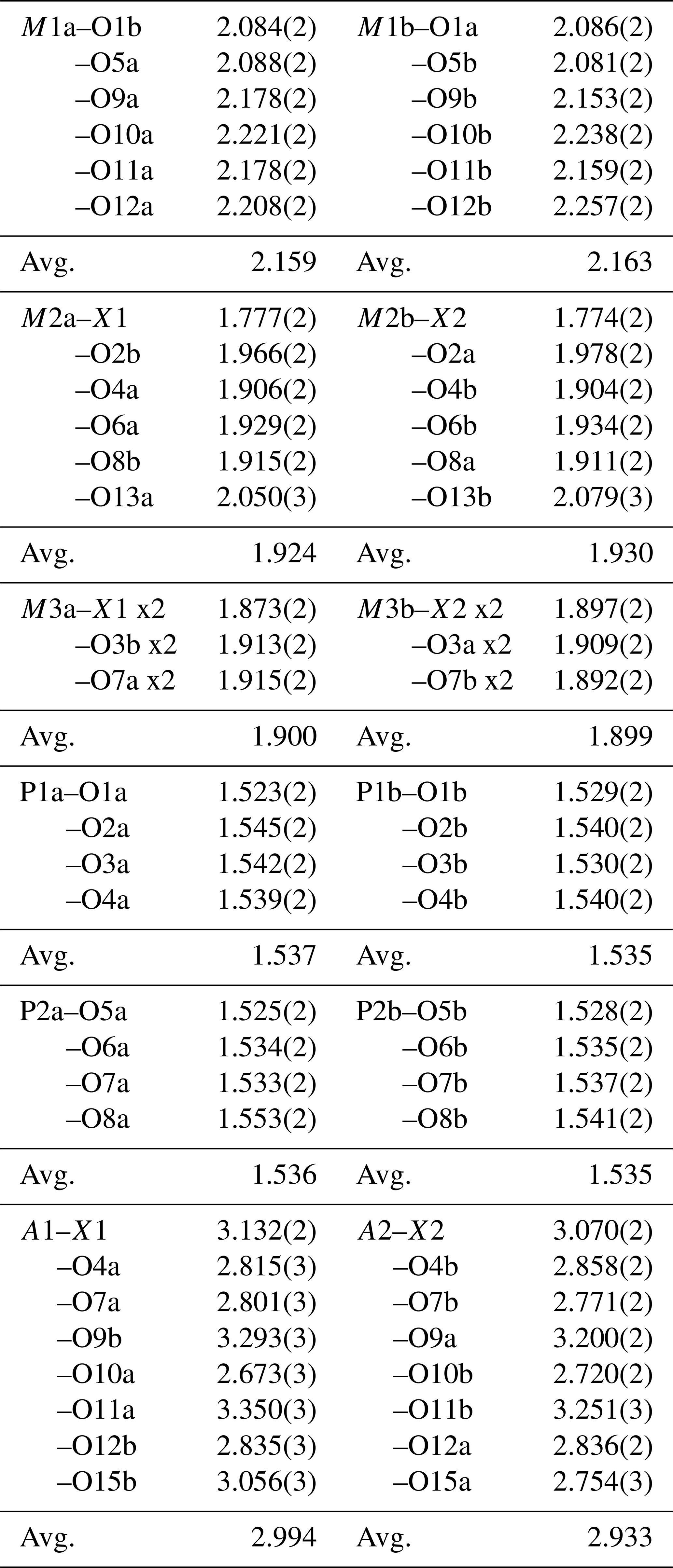
The crystal structure for fluor-rewitzerite can be described in terms of a structural unit plus an interstitial complex, using the terminology of Hawthorne (1992) and Hawthorne and Schindler (2008). The structural unit is a 3D anionic framework of corner-connected octahedra and tetrahedra, with formula [Mn2Al2Ti(PO4)4(OF)(H2O)10]1−, while the interstitial complex is [K(H2O)(H2O)4]1+, where the first H2O corresponds to transformer-type O15a, coordinated to K, and the four other H2O groups are involved only in H bonding. These comprise O14a, O14b, O15b and A1 in Tables 4 and 8. The structural unit is based on (001) heteropolyhedral layers at z = and , as shown in Fig. 5. The layers are built from [100] kröhnkite-type chains (Hawthorne, 1985) of four-member rings of corner-connected PO4 tetrahedra and M2O4X(H2O) octahedra. Each PO4 tetrahedron also shares a corner with M1O2(H2O)4 octahedra along [010]. The corner-shared linkages form eight-member rings of alternating octahedra and tetrahedra. Layers as shown in Fig. 5 are interconnected into an open 3D framework by corner-sharing of the M2O4X(H2O) octahedra with M3O4X2 octahedra located at z=0 and .
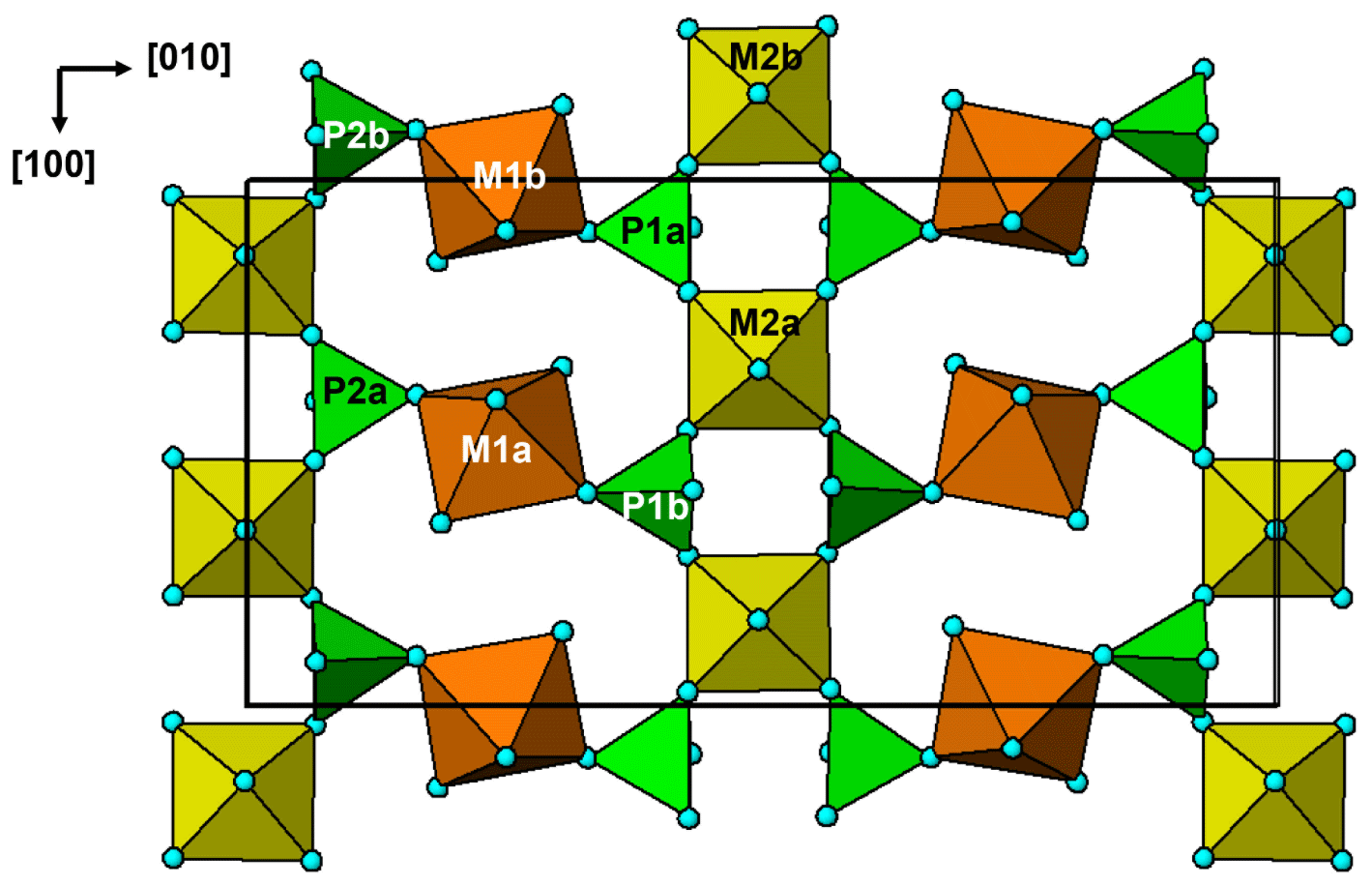
Figure 6The (001) section through the fluor-rewitzerite structure at z = 0. Dashed red lines on the left-hand side of the figure show H bonds, and solid blue lines on the right-hand side show bonding associated with the A1 and A2 sites. Arrows show [110] rows of regularly spaced H2O groups.
The interstitial complex is shown in Fig. 6. It corresponds to a (001) section through the structure centred at z = 0 or z = with a thickness of ∼ 2.7 Å. In addition to the interstitial constituents and the M3-centred octahedra, the section contains H2O groups that are coordinated to M1 cations (O10 and O12) and to M2 cations (O13). In-section H bonding is shown by dashed red lines on the left-hand side of Fig. 6, and in-section bonds to A1 and A2 are shown by the blue lines. Linear chains of regularly spaced water molecules, with a mean separation of ∼ 2.9 Å, are aligned along [110] as shown by the arrows in Fig. 6. The H bonding contributes to meeting the local bond valence requirements of undersaturated O anions shared between M-centred octahedra and PO4 tetrahedra. These are O1 to O8 in Table 4. Only O4a and O4b, shared between the M2-centred octahedra and PO4, are not acceptors for H bonds as their valence requirements are essentially met by M2 and P (see BVS in Table 4). In addition to the H bonds to O1 to O8, there are extensive H bonds between water molecules in the interstitial complex. The strongest is from O13 to O15, with an O13⋯O15 distance of ∼ 2.7 Å, shown in Fig. 6 and in Table 7. The O13a and O13b water molecules also form H bonds with three acceptor anions. In these cases, the H points towards a face X–O3–O7 of an M3-centred octahedron and forms H bonds with all three anions forming the face as shown in Fig. 6.
The major difference between the crystal structure of fluor-rewitzerite and that of the orthorhombic paulkerrite-group mineral benyacarite (Demartin et al., 1993) is an ordering of K and H2O at the A1 and A2 sites, whereas they are disordered at a single A site in benyacarite. It would be expected that the ordering of the positive K+ cation at a specific A site would be associated with a localised negative charge at the associated structural unit site. For the ideal end-member fluor-rewitzerite with a structural unit formula of [Mn2Al2Ti(PO4)4(OF)(H2O)10]1−, the local M2–X–M3-ordered groupings in the structural unit will be predominantly Al–F–Al and Al–O–Ti. The first is formally charge-balanced for octahedrally coordinated Al, but the latter will give rise to an undersaturated bridging O2− anion. It would be expected that the K+ would be associated with this configuration, and this appears to be borne out by the results in Tables 5 and 8. The K at the A2 site is coordinated to X2, which is shared between M2b containing dominant Ti and M3b containing dominant Al, whereas H2O at the X1 site is associated with M2a and M3a, both containing dominant Al. This ordering should be manifested in X2 being predominantly O2− and X1 being predominantly F−, but the BVS values in Table 4 do not reflect this. However, the BVS values for X1 and X2 are calculated from the mean positions of cations at the M2 and M3 sites, whereas these sites contain different cations, Al3+, Fe3+ and Ti4+, which may each be at different positions in the octahedra. The anisotropic displacement parameters (ADPs) for the M2 sites in Table 6 show elevated values for U33, corresponding to root-mean-square displacements of 0.17 Å. Displacements of individual cations by this magnitude will have large effects on the BVS values for the X sites. We tried refining the contents at these sites, but these anions are isoelectronic and the refinements were inconclusive. We also tried refinements in the non-centrosymmetric space group Pc, but the refinements were unstable (oscillating) and gave non-positive ADPs and large ranges for the P–O distances.
There are currently nine published paulkerrite-group minerals, which are distinguished compositionally in Fig. 4. The first three members to be characterised, paulkerrite (Peacor et al., 1984), mantienneite (Fransolet et al., 1984) and benyacarite (Demartin et al., 1993, 1997), were all described as having orthorhombic symmetry, space group Pbca, and this appeared to be confirmed from a single-crystal refinement for benyacarite. The new members pleysteinite and hochleitnerite (Grey et al., 2023c, d) were reported to be isostructural with benyacarite, based on laboratory-based single-crystal diffraction data. A single-crystal study on type paulkerrite using microfocus synchrotron diffraction data, however, revealed a small monoclinic distortion of the published unit cell (β = 90.33(3)°) with the crystal structure conforming to P21/c, a maximum non-isomorphous subgroup of Pbca (Grey et al., 2023b). Peacor et al. (1984) had already noted the poor diffracting quality of paulkerrite crystals and the “possibility of submicroscopic domains having a structure differing from the average”. We subsequently collected microfocus synchrotron diffraction data on crystals of pleysteinite and hochleitnerite and confirmed that these minerals are also monoclinic, P21/c (Rewitzer et al., 2024). The combination of a very small deviation of β from 90°, ubiquitous twinning and large mosaicity makes it difficult to determine the correct symmetry using conventional sealed-tube laboratory diffractometer data. Recent microfocus synchrotron data collected on the new paulkerrite-group minerals, rewitzerite (Grey et al., 2023a), hydroxylbenyacarite (Hochleitner et al., 2024) and macraeite (Grey et al., 2024), have confirmed the monoclinic character of these minerals. It is likely that the crystal structure of benyacarite is also of monoclinic symmetry. An indication of this comes from the very large displacement parameters reported for O15 (Demartin et al., 1993), with U ∼ 0.05 Å2 corresponding to a mean-square displacement of 0.22 Å. In going from the average Pbca structure to P21/c, the largest displacements occur for O15 in published monoclinic structures (Rewitzer et al., 2024). This is shown for fluor-rewitzerite in Table 8, where A1–O15b and A2–O15a distances differ by 0.30 Å, whereas all the other pairs of A–O distances differ by less than 0.1 Å. The large U values for O15 from the Pbca refinement of benyacarite thus suggest that the correct symmetry is monoclinic, at least in small domains.
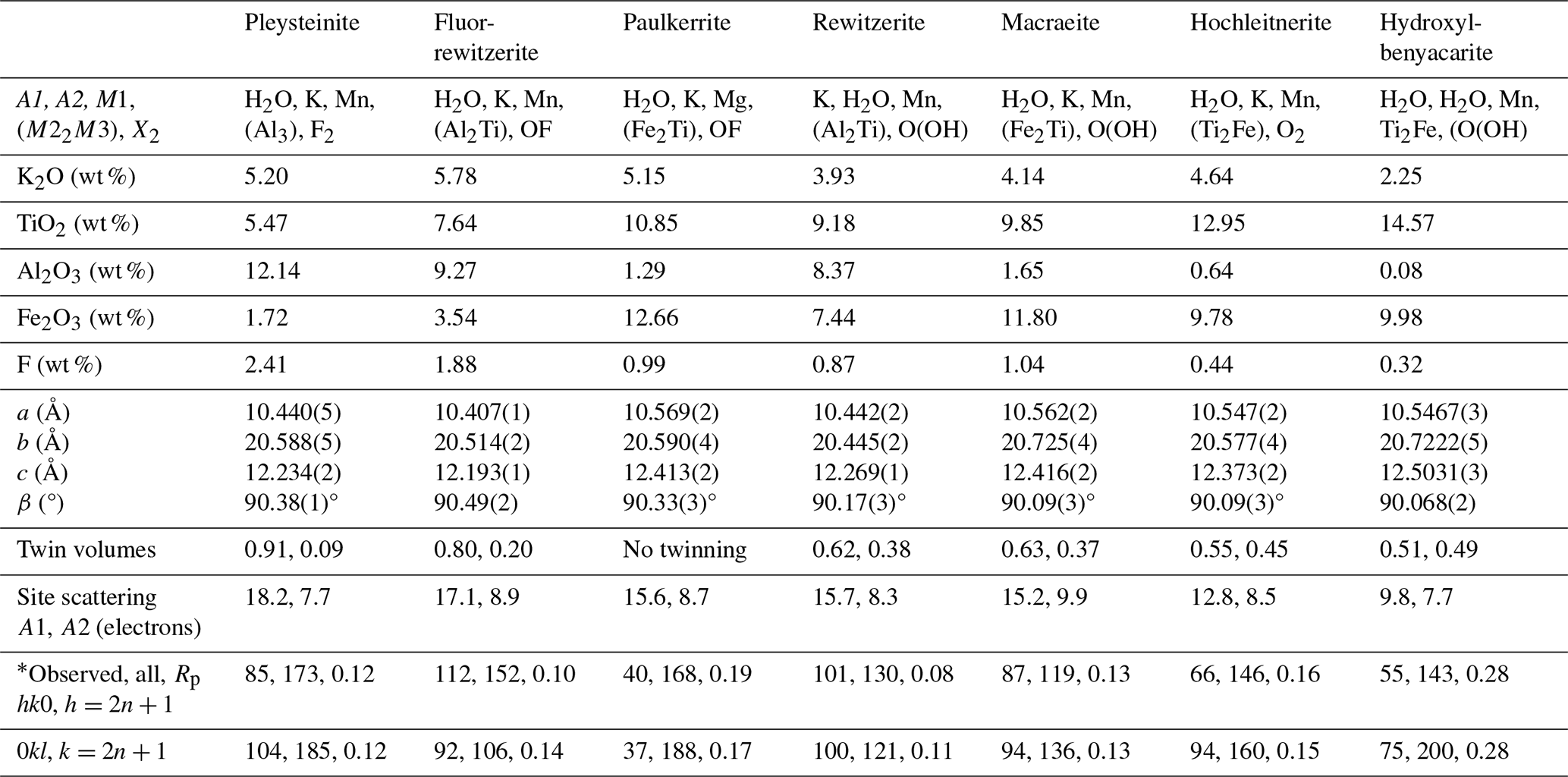
Table 9Crystal–chemical trends in monoclinic paulkerrite-group minerals, general formula A1A2M12M22M3(PO4)4X2(H2O)10⋅4H2O.
* Reflections forbidden in Pbca.
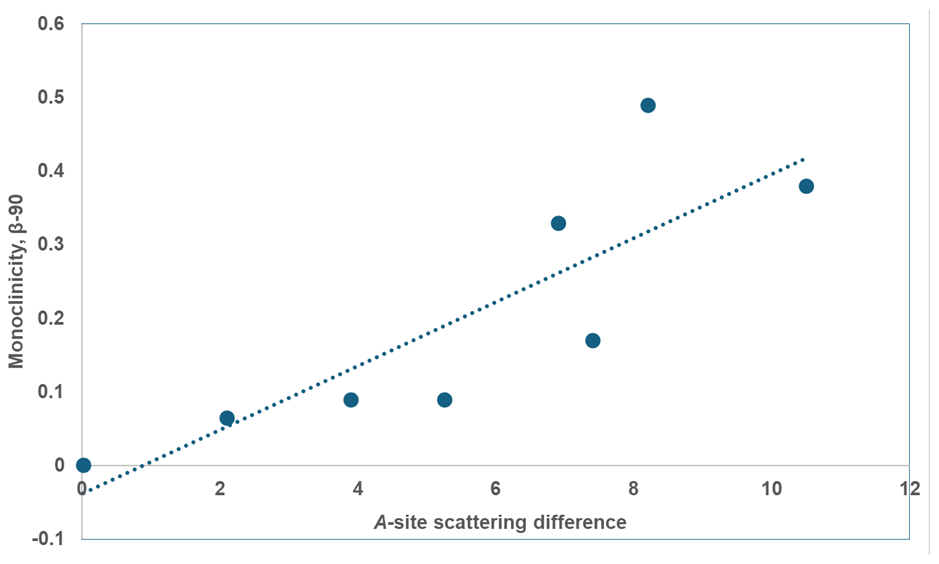
Figure 7Plot of A-site scattering difference vs. β-90 (°) for monoclinic paulkerrite-group minerals.
Some trends in the crystal chemistry of the seven monoclinic paulkerrite-group minerals are given in Table 9. The magnitude of the monoclinic distortion correlates positively with the difference in scattering between the A1 and A2 sites, that is, with the extent of ordering of K and H2O at the A sites. This is illustrated in Fig. 7 (R2=0.70). There is a correlation between the monoclinicity and the extent of twinning. The minerals with the greatest monoclinicity are untwinned or have a dominant twin individual, and decreasing monoclinicity is associated with a shift towards equal twin volumes. Also given in Table 9 is information on the reflections (hk0), , and (0kl), . These reflections are systematically absent in the diffraction patterns of the orthorhombic Pbca minerals due to the a and b glide planes. Their presence, as well as their fitting in the refinement, is the most direct evidence for the space group P21/c. The results for paulkerrite appear to be anomalous, with much lower numbers of these reflections, but this is because the reflection data are much weaker, with only 40 % of reflections having I>3σ(I) (Grey et al., 2023b) compared with ∼ 90 % for the other minerals. Aside from paulkerrite, there is a general trend from left to right (direction of decreasing monoclinicity and ordering) of smaller numbers of observed “forbidden” reflections and increasing partial R factors (Rp) in the fitting of these reflections. Hydroxylbenyacarite, which has only about half the K content of the other minerals, is the most extreme in terms of the smallest monoclinicity and A-site scattering difference, and it has the poorest fit to the forbidden reflection intensities. Reconstructed precession images of the diffraction data for this mineral show that the reflections (hk0), , and (0kl), , are diffuse with streaking parallel to b* (Hochleitner et al., 2024). The lengths of the diffuse streaks correspond to correlation lengths of only 1 to 2 nm, so the A-site ordering is restricted to domains on the scale of the unit cell. The results in Table 9 are consistent with a progressive decease in the size of the domains in which ordering of K and H2O at the A sites occurs, in the minerals from left to right in the table.
The general crystallographic trends shown in Table 9 correlate strongly with the mineral compositions. In particular, the decrease in A site scattering and associated smaller monoclinicity from left to right in Table 9 are associated with decreases in K, Al and F and with increases in Ti content. The variation in Fe3+ is less clear, but overall it increases from left to right, with the clear exception of paulkerrite. The elements Al, Fe3+, Ti and F are all associated with the octahedra at the M22M3 sites, suggesting that it is the distribution of compositions at these sites that determines the extent of monoclinic ordering. Figure 4 shows a distinct difference in the compositions of minerals that have Al > Fe at the M22M3 sites compared with those that have Fe > Al. The former all have M22M3 compositions in the range of Ti to (M3+-rich), whereas the latter have compositions in the range of Ti to M3+Ti2 (Ti-rich). Minerals in the Al-rich Al3 and Al2Ti phase fields (pleysteinite, fluor-rewitzerite and rewitzerite) have the highest degree of monoclinic ordering, whereas minerals in the Ti-rich Ti2Fe phase field have the lowest degree of ordering (hydroxylbenyacarite, hochleitnerite and benyacarite). The mineral mantienneite remains unclassified because the crystals are not of high-enough quality for a single-crystal structure determination. At present there are no known paulkerrite-group minerals with M22M3 compositions in the Fe3 or Ti2Al phase fields. Eventual discovery and characterisation of minerals in these two phase fields will help to further develop an understanding of the factors that determine the monoclinic ordering.
Crystallographic data for fluor-rewitzerite are available in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/ejm-36-541-2024-supplement.
IEG oversaw the research and wrote the paper; RH conducted initial characterisation studies on the specimen and identified it as a potential new species; WGM assisted in the diffraction data analysis; ARK measured the optical properties, Raman spectrum, powder XRD and crystal morphology; SB collected and processed the single-crystal diffraction data; and NCW and CMM developed the cryo-EMP analysis procedure and associated software.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We thank Cameron Davidson for preparing polished mounts for EMP analyses. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation detector.
This paper was edited by Sergey Krivovichev and reviewed by Gennaro Ventruti and one anonymous referee.
Aragao, D., Aishima, J., Cherukuvada, H., Clarken, R., Clift, M., Cowieson, N. P., Ericsson, D. J., Gee, C. L., Macedo, S., Mudie, N., Panjikar, S., Price, J. R., Riboldi-Tunnicliffe, A., Rostan, R., Williamson, R., and Caradoc-Davies, T. T.: MX2: a high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron, J. Synch. Radiat., 25, 885–891, 2018.
Bamberger, C. E., Begun, G. M., and MacDougall, C. S.: Raman spectroscopy of potassium titanates: Their synthesis, hydrolytic reactions and thermal stability, Appl. Spectrosc., 44, 31–37, 1990.
Demartin, F., Pilati, T., Gay, H. D., and Gramaccioli, C. M.: The crystal structure of a mineral related to paulkerrite, Z. Kristallogr., 208, 57–71, 1993.
Demartin, F., Gay, H. D., Gramaccioli, C. M., and Pilati, T.: Benyacarite, a new titanium-bearing phosphate mineral species from Cerro Blanco, Argentina, Can. Mineral., 35, 707–712, 1997.
Fransolet, A.-M., Oustriere, P., Fontan, F., and Pillard, F.: La mantiennéite, une novelle espèce minérale du gisement de vivianite d'Anloua, Cameroun, B. Mineral., 107, 737–744, 1984.
Gagné, O. C. and Hawthorne, F. C.: Comprehensive derivation of bond-valence parameters for ion pairs involving oxygen, Acta Crystallogr. B, 71, 562–578, 2015.
Grey, I. E., Hochleitner, R., Kampf, A. R., Boer, S., MacRae, C. M., Mumme, W. G., and Keck, E.: Rewitzerite, [K(H2O)]Mn2(Al2Ti)(PO4)4[O(OH)](H2O)10⋅4H2O, a new monoclinic paulkerrite-group mineral, from the Hagendorf Süd pegmatite, Oberpfalz, Bavaria, Germany, Mineral. Mag., 87, 830–838, 2023a.
Grey, I. E., Boer, S., MacRae, C. M., Wilson, N. C., Mumme, W. G., and Bosi, F.: Crystal chemistry of type paulkerrite and establishment of the paulkerrite group nomenclature, Eur. J. Mineral., 35, 909–919, 2023b.
Grey, I. E., Hochleitner, R., Rewitzer, C., Kampf, A. R., MacRae, C. M., Gable, R. W., Mumme, W. G., Keck, E., and Davidson, C.: Pleysteinite, [(H2O)0.5K0.5]2Mn2Al3(PO4)4F2(H2O)10⋅4H2O, the Al analogue of benyacarite, from the Hagendorf Süd pegmatite, Oberpfalz, Bavaria, Germany, Eur. J. Mineral., 35, 189–197, 2023c.
Grey, I. E., Keck, E., Kampf, A. R., MacRae, C. M., Gable, R. W., Mumme, W. G., Glenn, A. M., and Davidson, C.: Hochleitnerite, [K(H2O)]Mn2(Ti2Fe)(PO4)4O2(H2O)10⋅4H2O, a new paulkerrite-group mineral, from the Hagendorf-Süd pegmatite, Oberpfalz, Bavaria, Germany, Eur. J. Mineral., 35, 635–643, 2023d.
Grey, I. E., Rewitzer, C., Hochleitner, R., Kampf, A. R., Boer, S., Mumme, W. G., and Wilson, N. C.: Macraeite, [(H2O)K]Mn2(Fe2Ti)(PO4)4[O(OH)](H2O)10⋅4H2O, a new monoclinic paulkerrite-group mineral, from the Cubos-Mesquitela-Mangualde pegmatite, Portugal, Eur. J. Mineral., 36, 267–278, 2024.
Gunter, M. E., Bandli, B. R., Bloss, F. D., Evans, S. H., Su, S. C., and Weaver, R.: Results from a McCrone spindle stage short course, a new version of EXCALIBR, and how to build a spindle stage, Microscope, 52, 23–39, 2004.
Hawthorne, F. C.: Towards a structural classification of minerals: The viMivT2Φn minerals, Am. Mineral., 70, 455–473, 1985.
Hawthorne, F. C.: The role of OH and H2O in oxide and oxysalt minerals, Z. Kristallogr., 201, 41–68, 1992.
Hawthorne, F. C. and Schindler, M.: Understanding the weakly bonded constituents in oxysalt minerals, Z. Kristallogr., 223, 183–206, 2008.
Hochleitner, R., Rewitzer, C., Grey, I. E., Kampf, A. R., MacRae, C. M., Gable, R. W., and Mumme, W. G.: Hydroxylbenyacarite, (H2O)2Mn2(Ti2Fe)(PO4)4[O(OH)](H2O)10⋅4H2O, a new paulkerrite-group mineral from the El Criollo mine, Cordoba province, Argentina, Mineral. Mag., https://doi.org/10.1180/mgm.2024.16, online first, 2024.
Mandarino, J. A.: The Gladstone-Dale relationship: Part IV. The compatibility concept and its application, Can. Mineral., 19, 441–450, 1981.
Peacor, D. R., Dunn, P. J., and Simmons, W. B.: Paulkerrite a new titanium phosphate from Arizona, Mineral. Rec., 15, 303–306, 1984.
Petříček, V., Dušek, M., and Palatinus, L.: Crystallographic Computing System JANA2006: General features, Z. Kristallogr., 229, 345–352, 2014.
Rewitzer, C., Hochleitner, R., Grey, I. E., MacRae, C. M., Mumme, W. G., Boer, S., Kampf, A. R., and Gable, R. W.: Monoclinic pleysteinite and hochleitnerite from the Hagendorf Süd pegmatite. Synchrotron microfocus diffraction studies on paulkerrite-group minerals, Can. J. Mineral. Petrol., 62, 513–527, 2024.
Sejkora, J., Skoda, R., Ondrus, P., Beran, P., and Susser, C.: Mineralogy of phosphate accumulations in the Huber stock, Krasno ore district, Slavkovsky les area, Czech Republic, J. Czech Geol. Soc., 51, 103–147, 2006.
Sheldrick, G. M.: Crystal structure refinement with SHELXL, Acta Crystallogr. C, 71, 3–8, 2015.
Silva, F. L. R., Filho, A. A. A., Silva, M. B., Balzuweit, K., Bantiignies, J.-L., Caetano, E. W. S., Moreira, R. L., Freire, V. N., and Righi, A.: Polarized Raman, FTIR, and DFT study of Na2Ti3O7 microcrystals, J. Raman Spectrosc., 49, 535–548, 2018.
Tu, C.-S., Guo, A. R., Tao, R., Katiyar, R. S., Guo, R., and Bhalla, A. S.: Temperature dependent Raman scattering in KTiOPO4 and KTiOAsO4 single crystals, J. Appl. Phys., 79, 3235–3240, 1996.