the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 Jan 2022
| 25 Jan 2022
The influence of oxygen fugacity and chlorine on amphibole–liquid trace element partitioning at upper-mantle conditions
Massimo Tiepolo
Giulio Borghini
Antonio Langone
Patrizia Fumagalli
The recycling of volatile and incompatible elements through the entire Earth's history is one of the most important processes for the chemical evolution of the lithosphere. In this context, amphiboles are important hydrous minerals playing a key role in the chemical differentiation of the crust–mantle system. We carried out amphibole–liquid partitioning (D) experiments starting from a hydrous alkali basalt with variable chlorine content and doped in 37 trace elements of geochemical interest. Experiments were conducted with an end-loaded piston cylinder apparatus at a fixed pressure of 1.4 GPa, temperature from 1015 to 1050 ∘C, and at two different oxygen fugacity conditions (fO2; ΔFMQ 2.6 (log fO2 [experiment] − log fO2 [FMQ buffer]) and ΔFMQ 1.7). These conditions approach those of the Earth's upper mantle chemically metasomatised by subduction-derived agents that may occur in several geological settings. All runs show Ti-pargasites and kaersutites, either as individual crystals or replacing former clinopyroxenes, in equilibrium with alkali-rich (4 wt %–6 wt %) and silica-intermediate (52 wt %–59 wt %) melts. A comprehensive trace element dataset (LLEs – light lithophile elements, LILEs – light ion lithophile elements, REEs – rare Earth elements, HFSEs – high field strength elements, actinides, transition metals, chalcophiles) has been determined in glass and amphibole with a laser ablation inductively coupled plasma mass spectrometer (LA-ICP-MS). Although the melt polymerization has a major influence on Ds, we provide evidence that changes in the fO2 of both the system and Cl content in amphibole (up to 0.88 wt %) also affect the Ds of several trace elements and their ratios. In particular, at higher fO2 conditions lower D values for U and Mo are observed likely in relation to the low capability of amphibole to incorporate the more oxidized forms of these cations. The DDU ratio is positively correlated with the Cl content in amphibole, while the opposite behavior is shown by D ratios of other elements with the same charge and different ionic radius (e.g., Gd / Yb, Nb / Ta, and Pb / Ba). Dimensional modification of the amphibole structure in relation to the incorporation of Cl in the O(3) site are at the origin of the observed variations. General implications of our results are discussed.
- Article
(9183 KB) - Full-text XML
-
Supplement
(520 KB) - BibTeX
- EndNote





The partitioning of trace elements between minerals and silicate melts is a key topic in our understanding of how elements redistribute during Earth's evolution, from primordial times to present. The affinity of trace elements in the melt phase compared to their storage in minerals (i.e., incompatible behavior) acts as a geochemical filter playing a key role in the chemical differentiation of the core–mantle–crust system. In the last five decades many efforts have been dedicated to the determination of the mineral–liquid partition coefficient (D) for most of the trace element groups (e.g., light lithophile elements – LLEs, light ion lithophile elements – LILEs, rare Earth elements – REEs, high field strength elements – HFSEs, actinide, transition metals) mainly in major mantle-derived minerals like olivine, pyroxenes, garnet, and spinel and more typically crustal phases like feldspar, amphiboles, and micas, as well as accessory phases such as allanite, zircon, and rutile in more evolved petrologic systems (e.g., Wood and Blundy, 2014, and references therein).
The key parameters affecting D of trace elements are the initial composition of the system (e.g., acid vs. basic and volatile-bearing vs. anhydrous chemical systems) together with its intensive parameters – i.e., pressure and temperature (P−T) conditions (e.g., Adam and Green, 2003; Dalpé and Baker, 2000; Green et al., 2014). The oxygen fugacity conditions (i.e., activity of oxygen – fO2) is a further factor able to affect the trace element incorporation within a specific mineral during its crystallization (e.g., Dalpé and Baker, 2000). Together with the H2O contents, the fO2 also exerts a strong influence on the physical behavior of magmas, the phase relations, and mineral chemistry (Feig et al., 2010, 2006; Foley, 2011; Jenner et al., 2010; Sisson et al., 2005; Stamper et al., 2014; Ulmer et al., 2018). How the fO2 affects the Ds of trace elements is more evident by considering minerals with complex and more versatile crystal structure, such as those of amphiboles and micas (Ferraris and Ivaldi, 2002; Oberti et al., 2007). This is mainly due to their greater ability to counterbalance charge imbalance imposed by the incorporation of trace elements with different oxidation states (e.g., V, Ni, Mo, U) and to accommodate the incorporation of cations with both small (e.g., [6]Be.45 Å) and large (e.g., [8]Ba.42 Å) ionic radii.
Amphiboles are the most important hydrous minerals crystallizing in mafic/ultramafic systems because they can form either at the expense of mantle pyroxenes or during hydrous magma crystallization at depths less than 100 km. Amphibole stability includes upper-mantle/lower-crust depths (e.g., Coltorti et al., 2004; Davidson et al., 2007; Müntener and Ulmer, 2006; Pirard and Hermann, 2015b; Smith, 2014) where they act as the main repository for several trace elements of geochemical interest (Tiepolo et al., 2000a). The complex and variable crystal chemistry of high-T magmatic amphibole is affected by several chemical mechanisms able to modify the major element distribution within the amphibole's crystallographic sites and to exert a major control of the amphibole's volatile budget. The deprotonation in the O(3) site (i.e., the loss of H+ from the hydroxyl anion OH−), the so-called oxo-component, can be balanced by crystal chemical mechanisms involving either Ti4+ or Fe3+ (King et al., 1999; Oberti et al., 2007; Popp et al., 2006, 1995), and this crystallographic process may induce variations in the Ds. For instance, the Ti4+ incorporation in the M(1) site of the amphibole (instead of Mg2+ or Fe2+) influences the size of the M(1) octahedron and, consequently, induces variation in the <M(1)-O(3)> distance, thus affecting the DDTa ratios (Tiepolo et al., 2000b, 2007). In the O(3) site, the OH− can be substituted by Cl− with implications for the whole crystal structure of the amphibole itself (Makino et al., 1993; Oberti et al., 1993). The higher ionic radii of Cl− with respect to other volatile species entering in the O(3) site affect the <M(1)-O(3)> and <M(3)-O(3)> distances and the size of the A site of the amphiboles. These changes could affect the ratios between Ds of trace elements with different ionic radii and entering in the same site. The Cl-rich amphiboles usually originate from Cl-rich fluids rather than from Cl-rich melts (e.g., Giesting and Filiberto, 2016, and reference therein). However, a few experimental works have dealt with the genesis of Cl-rich amphibole in magmatic-equivalent systems (Campanaro and Jenkins, 2017; Chan et al., 2016; Jenkins, 2019), and they provided evidence that both Fe and K contents in amphiboles strongly influence the Cl incorporation.
Another parameter that can potentially affect the Ds of trace elements is the oxidation state, which is known to be heterogeneous in the Earth's mantle (Foley, 2011; McCammon, 2005). It is expected to be strongly reduced in the deep lower mantle (lower than 5 log units below the fayalite/ferrosilite–magnetite–quartz – FMQ – buffer) and variably oxidized in the upper mantle (between −4 and +2 log units below the FMQ buffer). The broad fO2 variability in the upper mantle likely reflects the deep injection of surficial and mostly oxidized materials by recycling processes active in convergent margins (e.g., Cannaò and Malaspina, 2018, and references therein). Dalpé and Baker (2000) experimentally determined the effect of fO2 on trace element partitioning between Ca-amphibole and basaltic melt. They demonstrate that changes in fO2 affect the Ds of several trace elements and modify the amphibole's crystal structure. This study was limited to Ds of LILEs, HFSEs, and REEs. Determination of Ds of other important element groups, such as LLEs, transition metals, and actinides, at different fO2 is still lacking, and it represents a key mandatory aspect in order to fully investigate Earth's processes occurring at upper-mantle conditions (e.g., Nandedkar et al., 2016; Tiepolo et al., 2000a, 2007).
In this contribution, we provide a new set of experimentally determined Ds for a large number of trace elements aiming to better understand the effect of fO2 and Cl on trace element partitioning between amphibole and hydrous basaltic melt at lower-crust and upper-mantle conditions. We experimentally derive a comprehensive set of Ds obtained in Cl-poor and Cl-rich systems at both low and high fO2 conditions: ΔFMQ 2.6 (log fO2 [experiment] − log fO2 [FMQ buffer]) and ΔFMQ .7, respectively. These conditions fit with the fO2 estimated values of the Earth's upper mantle (ΔFMQ ) based on oxygen thermobarometry measurements on spinel-bearing peridotites (e.g., Frost and McCammon, 2008) and simulate hydrous melt-related metasomatism of Earth's upper mantle occurring in several geological settings (e.g., supra-subduction and intraplate settings).
2.1 Starting material
We reproduced the composition of the initial hydrous basalt in the chemical system K2O-Na2O-CaO-MgO-FeO-Al2O3-TiO2-SiO2-H2O starting from a mix of oxides and carbonates (for more information see Tiepolo et al., 2000a). The carbonate component was eliminated by sintering the mixture for 1 h at 930 ∘C, whereas, to avoid Fe loss, iron was added to the carbonate–oxide mixture after the sintering process as Fe0 + Fe2O3. In order to promote the crystallization of amphibole, a different amount of distilled water (nominally from 5.0 % to 13.2 % of the total volume) was added with a microliter syringe in each single experimental charge. The starting material was doped with a mixture of 33 trace elements of geological relevance (REEs, LLEs, LILEs, HFSEs, transition metals, and actinides) as already described in Tiepolo et al. (2000a). A 1000 ppm ICP standard solution was used as source for As and Sb (As in 2 % HNO3; Sb in 2 % HCl) and added to the distilled water used to hydrate the initial system. Table 1 reports the major and trace element composition of the starting material.
Table 1Concentrations of major and trace elements in the starting glass (liquid) at 1300 ∘C (exp 18-14C).
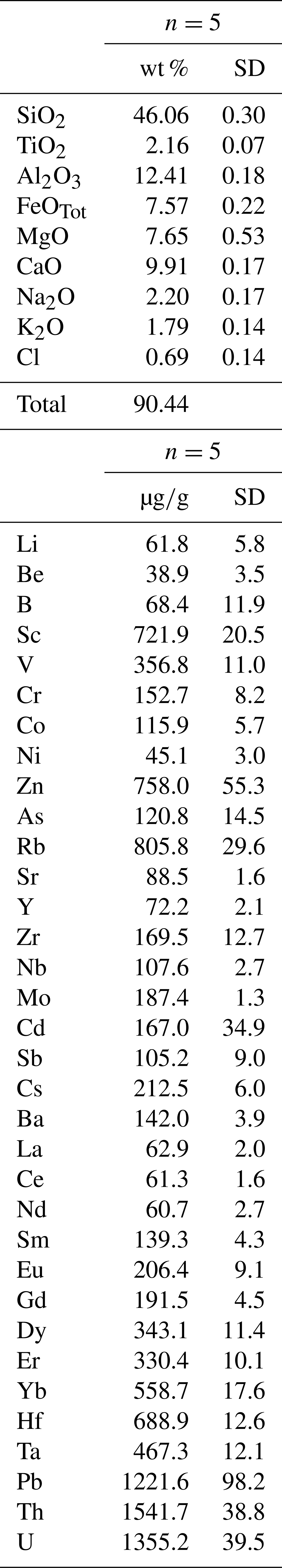
2.2 Experimental techniques
Experiments were performed at the Dipartimento di Scienze della Terra (University of Milan, Italy) at the fixed pressure of 1.4 GPa and T ranging from 1015 to 1300 ∘C (Table 2) in an end-loaded piston cylinder apparatus using MgO–Pyrex–Talc assembly. For all experimental runs, an initial pressure of 0.25 GPa was applied, and the sample was first heated to 400 ∘C for 20 min in order to soften the Pyrex. Then pressure was raised to the experimental value before reaching the T of 1300 ∘C. After 1 h at 1300 ∘C, when the starting basalt is at super-liquidus conditions (see run 47-N 18-14C; Table 1), experimental runs were cooled down to the equilibrium T (ranging from 1050 to 1015 ∘C), following the T-time path shown in Fig. 1b (with a ramp of 0.5 ∘C/min). This procedure optimizes the growth of small numbers of large and (possibly) unzoned crystals of amphibole. Initial quench rates of 90 ∘C/s were attained by cutting off the power after 12 hours at the target T (Fig. 1b). The thermocouple tip was separated from the Pt capsule by a 0.6 mm thick hard alumina disk. K- and S-type thermocouples were used to measure the T of the experiments and were considered to be accurate to ±5 ∘C. According to piston cylinder calibration, pressure uncertainty is assumed to be ±3 %. Experimental capsules were then embedded in epoxy resin and polished using non-aqueous lubricants for subsequent analytical analyses. Table 2 contains a summary of experimental conditions, glass and amphibole compositions, and coexisting phases.
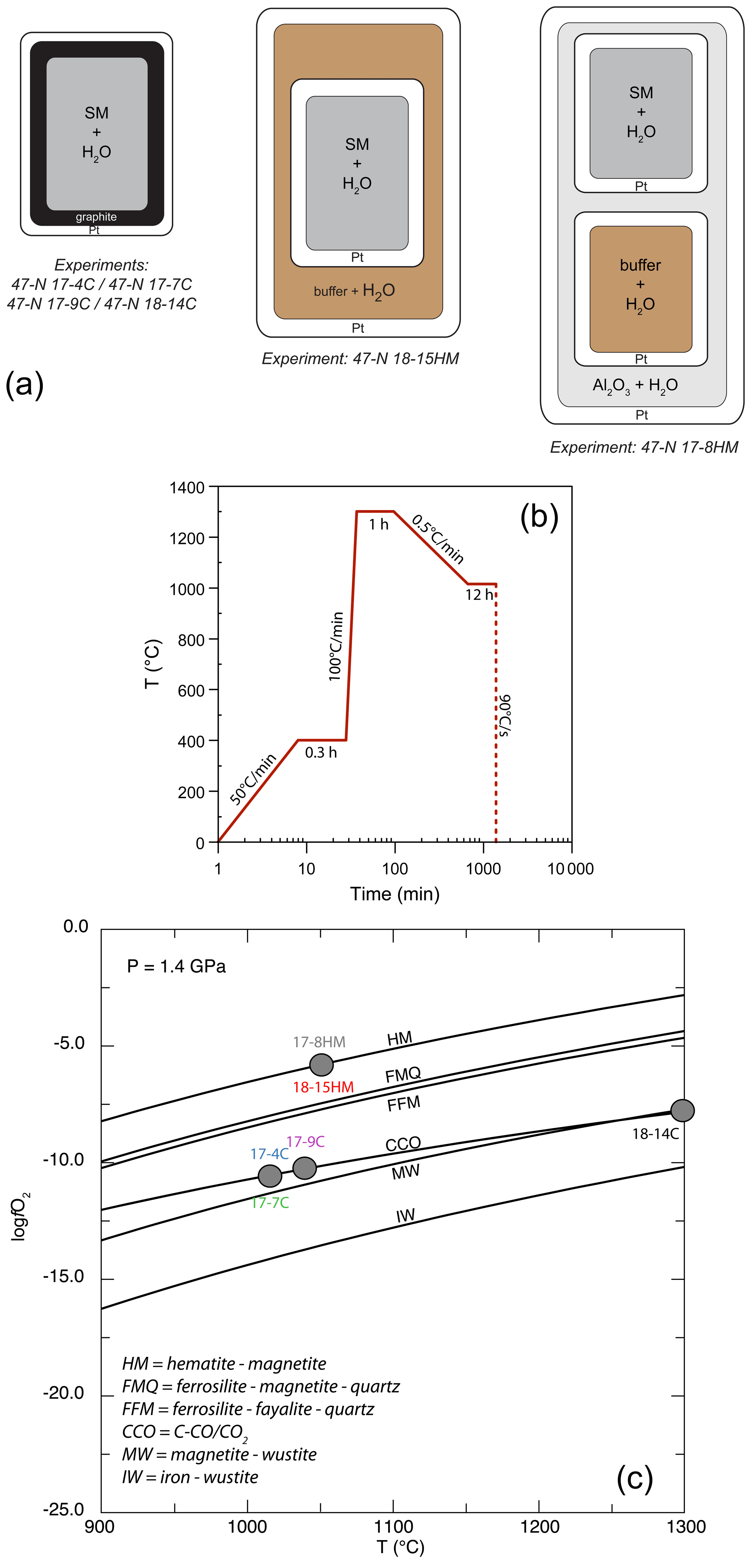
Figure 1(a) Single-, double-, and triple-capsule design used in crystallization experiments; SM is “starting material”. (b) Temperature vs. time path of the experiments. (c) Temperature vs. log fO2 diagram at fixed pressure of 1.4 GPa reporting the different experimental runs (see Table 2 for more information). The solid lines represent fO2 conditions by adopting different buffers. Calculating fO2 was achieved using the Perple_X software (Connolly, 2005).
Table 2Summary of experimental conditions and run products.
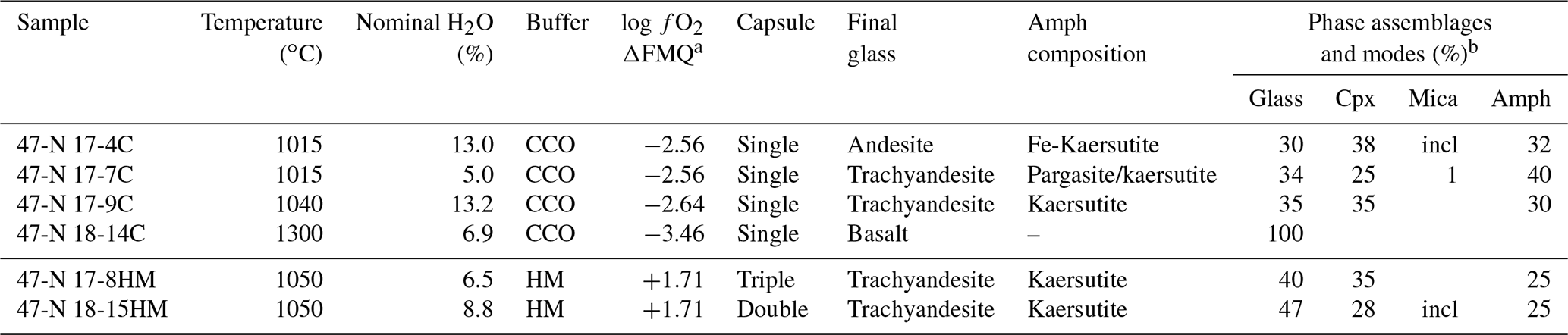
a Calculated from Fig. 1. Single capsule uses the graphite sleeve. Amph: amphibole – Cpx: clinopyroxene – incl: inclusion in amphibole. b Modal amount of mineral calculated via mass balance (see Cannaò et al., 2022). Ilmenite is present as accessory phase in all experiments.
2.2.1 Single-capsule experiments
For the experiments 47-N 17-7C/9C and 14C, approximately 12 mg of starting material was loaded into a graphite inner capsule (outer diameter – OD – ∼ 2.8 mm) and then seal-welded into an outer Pt capsule (OD = 3.0 mm, length about 8–10 mm; Fig. 1a). Graphite is used to isolate the sample from Pt capsule and avoid Fe loss (Johannes and Bode, 1978). The graphite–Pt assembly maintained the oxygen fugacity during the experimental run close to the C-CO–CO2 equilibrium (CCO; Ulmer and Luth, 1991), which at our run conditions is about FMQ −2.6 (Fig. 1c; Table 2). In the experiment 47-N 17-4C a similar capsule design was used even if its dimensions were bigger (OD = 5.0 mm, ID = 4.5 mm). In this case, about 30 mg of starting material was loaded into the graphite inner capsule.
2.2.2 Experiments with double and triple capsule
Experiments with the double- (Fig. 2c) and triple-capsule techniques allowed for the controlling of the oxygen fugacity fO2 of the system during experiments (run 47-N 17-8HM and 18-15HM; Fig. 1a). We adopted the hematite–magnetite (HM) buffer that enforces a higher redox condition with respect to the CCO experiment setting (FMQ + 1.71; Fig. 1c; Table 2). In order to minimize the Fe loss from the starting material, Fe preconditioning of Pt capsule (OD = 2.3 mm, ID = 2.0 mm) has been performed by using a gas mixing vertical furnace at the Dipartimento di Scienze della Terra (University of Milan, Italy). The Pt capsules were put in an iridium pot and filled in- and outside with powder of natural basalt (with similar FeO content of the starting material). The material was melted at 1300 ∘C for 24 h controlling the fO2 (at a value close to FMQ + 2) of the furnace using a CO-CO2 gas mix fixed by using a Tylan FC-260 flowmeters-group controller. A solution of HF and HClO4 (ratio of 3:1) was used to dissolve the glass product and to clean the capsules in order to avoid any contamination with the pre-saturation material. In addition to Fe, trace elements like Pb, Co, Ni, As, and Sb may form alloys with the Pt capsule (Adam and Green, 2006; see Supplement Fig. S1).
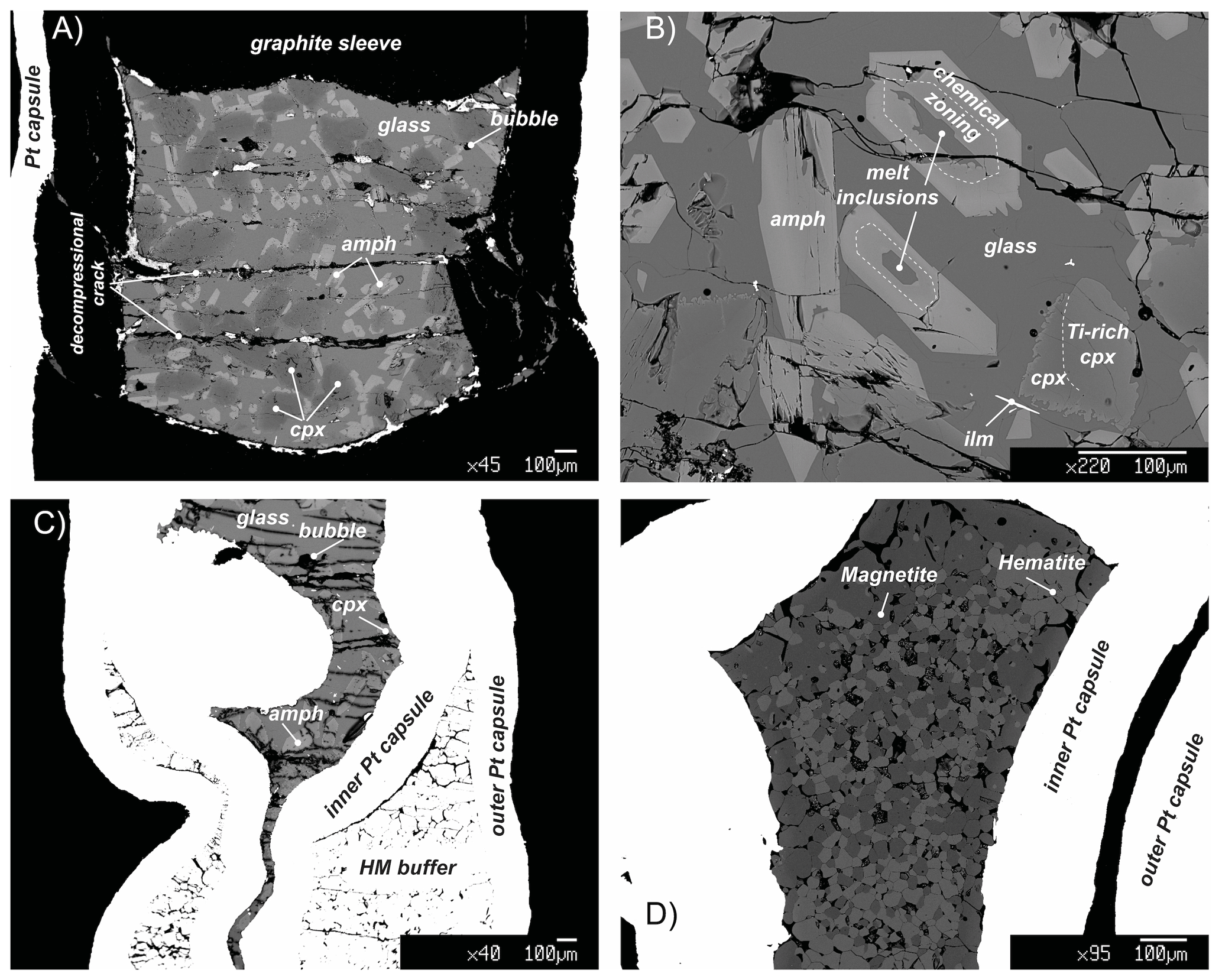
Figure 2Representative back-scattered electron (BSE) images showing the representative textures in key experiments. (a) Panoramic view of the capsule of the run 17-9C: amphibole (amph) shows a rather polygonal habit and is crystallized from the residual melt (glass) reacting with the former clinopyroxene (cpx). (b) Texture of experiment 17-4C showing amphibole (amph) replacing clinopyroxene (cpx). Amphibole includes small pockets of glass that reacts with the amphibole creating light chemical zoning. During the cooling, clinopyroxene grains react with the surrounding melt as evidenced by the corroded grain boundaries (ilm: ilmenite). (c) BSE overview image of the run 18-15HM (double-capsule design). (d) BSE image of the hematite–magnetite (HM) buffer of the run 17-8HM (triple-capsule design).
In the double-capsule experiment (47-N 18-15HM), a seal-welded inner (Fe-bearing) Pt capsule (OD = 2.3 mm) containing about 10 mg of starting material (plus As–Sb doped distilled H2O) was placed inside an outer-welded Pt capsule (OD = 3.0 mm) filled with the buffering assemblage (hematite + ∼ 30 wt % of distilled H2O). In the triple-capsule experiment (47-N 17-8HM), a seal-welded inner (Fe-rich) Pt capsule (OD = 2.3 mm) containing about 10 mg of starting material (plus As–Sb doped distilled H2O) and a second seal-welded inner Pt capsule (OD = 2.3 mm) with the buffering assemblage (hematite + ∼ 30 wt % of distilled H2O) were placed inside an outer-welded Pt capsule (OD = 3.0 mm) filled with a medium material (Al2O3+ distilled H2O). According to Matjuschkin et al. (2015), this capsule design is more accurate in monitoring and controlling the fO2 in high-pressure piston cylinder experiments.
In both experimental setups, the intrinsic fugacity of the end-loaded piston cylinder apparatus coupled with the use of talc in the assembly, leads to the partial reduction of the hematite into magnetite (HM buffer) during the run (e.g., Ulmer and Luth, 1991). As long as hematite + magnetite + H2O are present, this buffer fixes the fugacity of O2 and H2 through the following reactions:
and
which can be condensed to the following reaction:
Within the inner capsule, the dissociation of water at high T will produce oxygen and hydrogen according to Reaction (R2). As the Pt capsule is partially permeable to the hydrogen, the fugacity of the H2 in the inner capsule is expected to be the same as the fugacity of H2 of the outer capsule (Chou et al., 1978). Indirectly, also the fO2 will be fixed by the HM buffer (Luth, 1989). The persistence of the buffering assemblage (i.e., coexistence of hematite and magnetite) is verified by means of scanning electron microscopy (Fig. 2d).
2.3 Analytical techniques
Run products were inspected by back-scattered electron (BSE) images and microanalyses for the major element mineral compositions performed using a JEOL JXA-8200 SuperProbe equipped with five wavelength-dispersive spectrometers (WDSs) and one energy-dispersive spectrometer (EDS) at the Dipartimento di Scienze della Terra, University of Milan (Italy). The standards used were from natural materials: Mg on olivine, Fe on fayalite, Na on omphacite, Ti on ilmenite, Mn on rhodonite, K on K-feldspar, Al and Ca on anorthite, Si on wollastonite, Cr on metallic/pure Cr, Cl on scapolite, and Ni on niccolite. Analyses on mineral phases were performed using 1 µm beam size (up to 3 µm for the glass and mica) and beam conditions of 15 kV and 5 nA. Counting time was 30 s for peak and 10 s for the background.
Trace element mineral composition was determined by a laser ablation inductively coupled plasma mass spectrometer (LA-ICP-MS) at the IGG-CNR-UOS, Pavia (Italy). The instrument couples a 213 nm Nd:YAG laser microprobe (New Wave research) to a quadrupole ICP-MS system (DRC-e from PerkimElmer). Beam sizes were chosen as large as possible (up to 60 µm) to lower the limits of detection. The ablation system was operated at 10 Hz of repetition rate and with a fluence of ∼ 5.0 J/cm2. The NIST SRM 612 was used as external standard, whereas 43Ca or 29Si was selected as an internal standard, depending on the analyzed mineral. The USGS reference sample BCR-2G was analyzed as unknown in each analytical runs for quality control. Accuracy and precision are better than 10 % and 5 %, respectively. Data reduction was carried out using the Glitter software package (Griffin et al., 2008).
3.1 Phase assemblages and texture
Run products together with experimental details are summarized in Table 2. Isothermal run 47-N 18-14C contains only basaltic glass, confirming that the selected hydrous basalt is at super-liquidus condition at 1.4 GPa and 1300 ∘C. Run products in cooled experiments are glass, clinopyroxene, amphibole and mica (mica only in experiments 17-4C and 17-7C; Table 2). Ilmenite is present as accessory phase. Amphibole is the only phase stable with the glass in all the cooled runs at both CCO and HM oxygen fugacity conditions (Table 2). The amount of residual glass varies between 30 % and 47 %. Clinopyroxene is 100 to 300 µm in size (Fig. 2a), and it shows irregular habits and lobate, curvilinear boundaries, suggesting disequilibrium with the surrounding glass (Fig. 2b). Amphibole occurs as single crystal with euhedral habitus showing sharp and straight boundaries against the glass (Fig. 2a, b), but it can also replace the former clinopyroxene crystals and may include glass (Fig. 2b). Amphibole crystals range from 50 to 100 µm in size; in place, they are elongated crystals and may reach up to 350 µm. In some experimental runs, mica grains are included into the amphibole (17-4C and 17-7C) or dispersed in the glass showing irregular boundaries (only in experiment 17-7C).
Due to the high TiO2 content of the starting material (Table 1) required to stabilize amphibole crystallization at high T, small acicular grains of ilmenite form randomly along the entire capsule (Fig. 2). Glass is overall limpid and does not show quenched crystals (Fig. 2).
Phase assemblages and textures are similar between the different experimental runs with no significant difference as a function of experimental setups (i.e., single- vs. double-/triple-capsule techniques) and the fixed fO2.
3.2 Glass and mineral chemistry
Major and trace element compositions of glasses and amphiboles are reported in Tables 3 and 4, respectively; major element compositions of clinopyroxenes and micas are provided in the Supplement Table S1.
Table 3Glass compositions (major elements in wt % and trace elements in ).

* SD – standard deviation.
Table 4Amphibole compositions (major elements in wt % and trace elements in ).

* SD – standard deviation. bdl: below detection limit.
The glass compositions mainly fall in the trachyandesite/andesite fields (Fig. 3a). Glass chemical variations relative to the initial composition reflect the crystallization of clinopyroxene and amphibole, as revealed by the SiO2 and MgO vs. CaO and TiO2 correlations (Fig. 4). Glasses show moderate Cl contents (from 0.74 wt % to 2.40 wt %; Table 3) in relation to the Sb doping ICP standard solution (HCl 2 %; see Sect. 2.1). Within each single experimental charge, the trace element variations (e.g., REEs) in glasses from the top to the bottom of the capsule, as well as close to and far from the amphibole crystals, are almost negligible (Fig. 5a and b).

Figure 3Major element compositions of glass (a), clinopyroxene (b), and amphibole (c). In (a) glass compositions represent average values of data normalized to 100 wt %. When not visible, the error bar is smaller than the symbol.
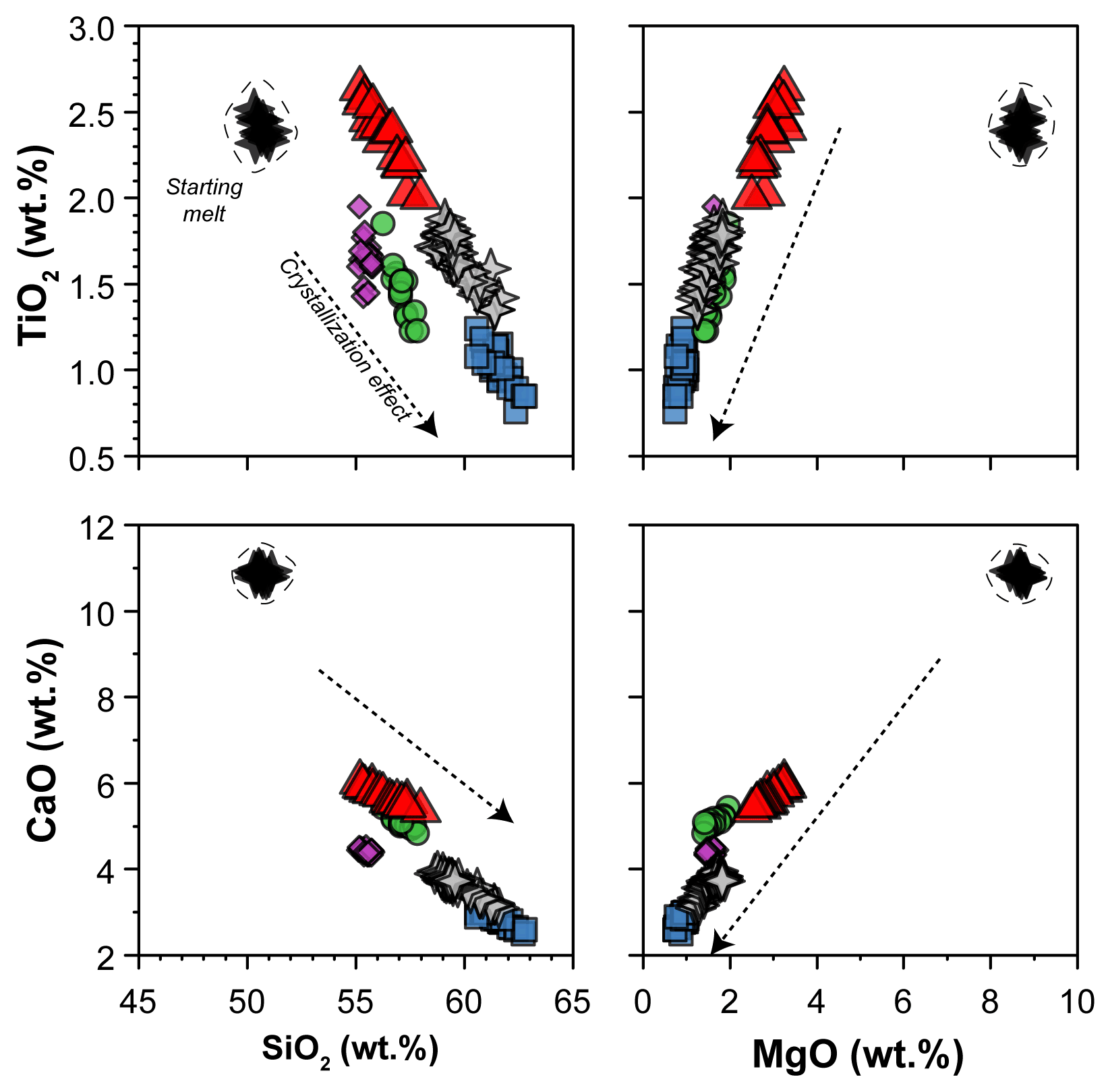
Figure 4SiO2 and MgO vs. CaO and TiO2 contents (in wt %) of glasses (normalized anhydrous to 100 wt %) after crystallization experiments. The black stars (within the dashed field) show the composition of the starting material. The arrows represent the effect of clinopyroxene plus amphibole crystallization on the composition of the evolving melts during crystallization experiments. Symbols are as in Fig. 3.
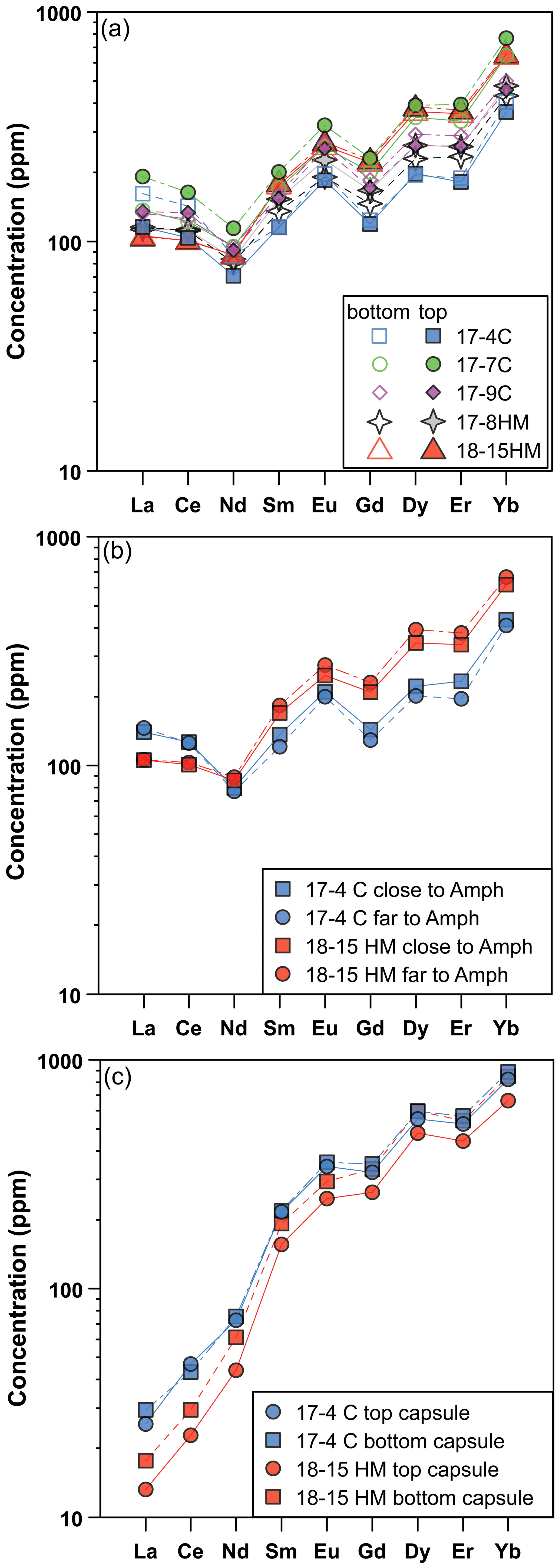
Figure 5Rare Earth element (REE) concentrations (in ppm) of glass (a, b) and amphibole (c) in experiments as a function of their location in the experimental charges (at top and bottom, a and c) and the REE composition of the glass close and far with respect to the amphibole grains (b). These patterns reveal rather homogeneous REE concentrations in glass and amphibole from the experimental charges. In (b) and (c) are shown experiments 17-4C and 18-15HM as representative of low and high fO2 conditions, respectively.
The clinopyroxenes have a composition in between diopside and augite in all experiments (Fig. 3b). The most enstatite-rich composition belongs to the clinopyroxene showing partial reaction with the glass, suggesting that these grains were the first to crystallize (see Fig. 6). Few clinopyroxenes show core-to-rim chemical variation due to the continuous equilibration with the melt during cooling towards amphibole stability (Figs. 2d and 6). With decreasing T, clinopyroxene records SiO2 and MgO contents decreasing coupled with TiO2 and Al2O3 contents increasing (Fig. 6). Noteworthy, this chemical evolution ends with the complete replacement of clinopyroxene by amphibole as the liquidus phase of evolved melts (Fig. 6).
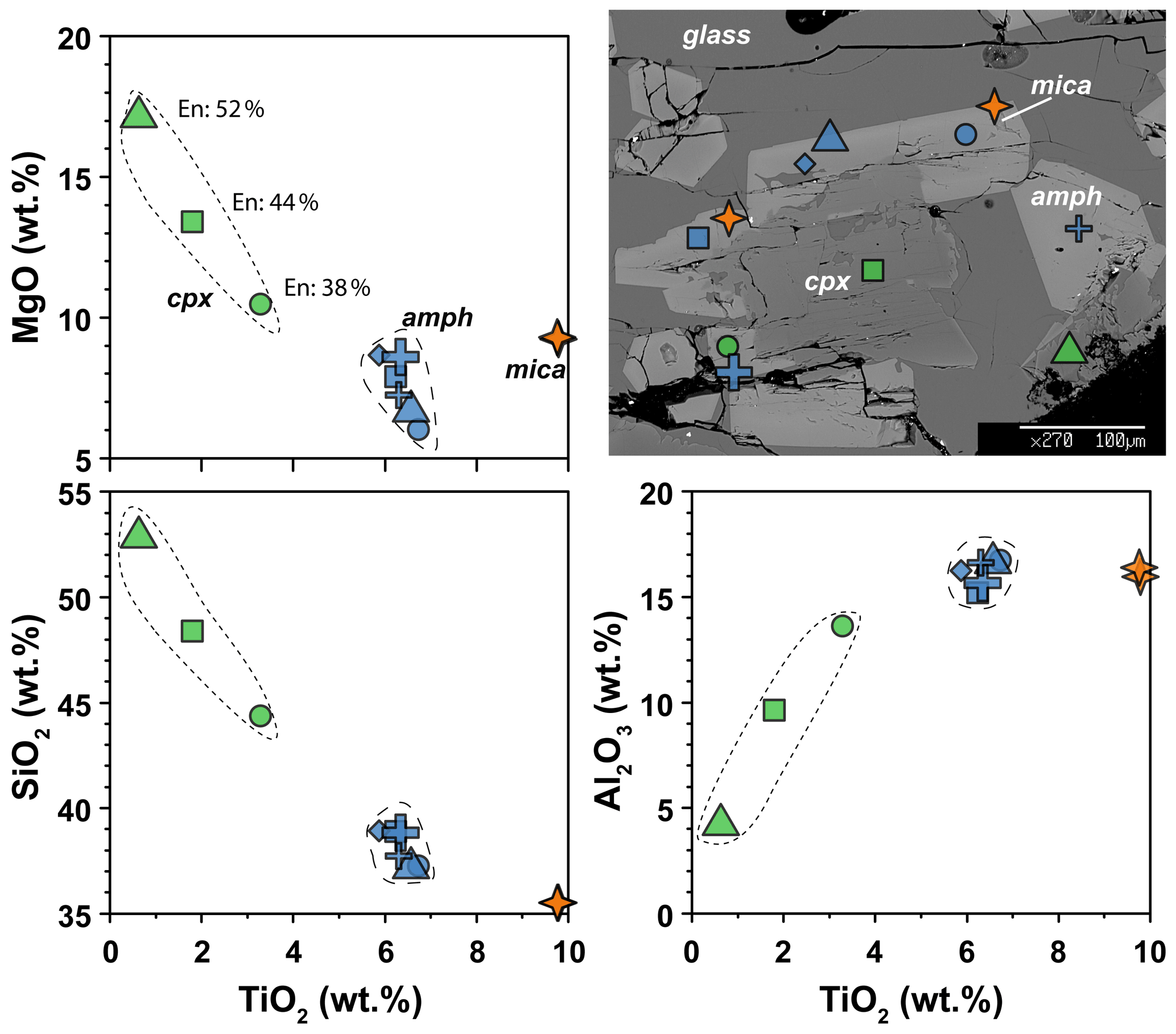
Figure 6Major element compositions of amphibole, clinopyroxene, and mica in experiment 17-4C. BSE image shows the representative texture and the location of the chemical analyses. En: enstatite component in clinopyroxene (expressed in vol %).
Amphiboles formulae were calculated in agreement with the protocol for EMPA data proposed by Li et al. (2020). These authors developed a machine-learning algorithm, principal component regression, based on references datasets, to establish a new calculation method for amphibole formula, which could solve the potential closure and multicollinearity in mineral chemical data and successfully produce a reasonable cation-/anion-assigned formula from EMPA data. This method allows us also to calculate the Fe3+ Fe2+ ratio and H2O content of amphibole. Amphiboles span in composition from kaersutite to Fe-kaersutite (Fig. 3c), with the exception for some pargasites and Fe-pargasite (following the classification of Leake et al., 1997) from the experiment 17-7C having the lowest Ti (< 0.5 a.p.f.u. – atom per formula unit). Amphiboles crystallized at high fO2 are characterized by Mg# (0.67–0.69) higher than those crystallized at lower fO2 (Mg# = 0.41 to 0.53; Fig. 3c). This is likely related to a higher Fe loss during the experimental run, although the effect of the increase in the fO2 of the experiment cannot be completely discharged. On average, amphiboles crystallized at high fO2 have higher Fe3+ Fetot (on average 0.106 to 0.161) than amphibole crystallized at low fO2 (on average 0.056 to 0.082), except for the amphibole in experiment 17-7C that shows high Fe3+ (on average 0.116) to compensate for the low Ti (a.p.f.u.) content. The Cl content in our amphiboles is unusually high, ranging on average from 0.31 wt % to 0.88 wt % (i.e., 0.080 to 0.232 a.p.f.u.) with no significant correlation with the fO2 conditions of the experimental runs. The complete amphibole recalculation data are presented in the Supplement Table S2.
All amphiboles show minor chemical variations (Fig. 3c), suggesting that at different experimental conditions its crystallization occurred very close to the liquidus. Major element homogeneity was checked by EMPA traverses within single amphibole grains (rim–core–rim, e.g., Fig. 6) and by comparing amphibole analyses collected in each run. Little chemical zoning of amphibole is observed only close to melt inclusions (Fig. 2b; e.g., see blue diamond and triangle in Fig. 6). These grains were thus excluded from the in situ trace element analyses. The REE concentrations in amphibole from the top to the bottom of the experimental charge show very little variation in terms of absolute concentrations (Fig. 5c). Intragrain trace element homogeneity was not evaluated due to the relatively small dimension of the single amphibole crystals that did not allow multiple LA-ICP-MS spot analyses. We filtered all the laser analyses on amphibole in order to exclude any potential contamination from melt inclusions by inspecting the concentration of highly incompatible elements such as Cs (e.g., Adam and Green, 2003).
In the experimental runs 17-4C and 17-7C, few mica grains occur as small crystals enclosed in amphibole crystals (Fig. 6), as well as single phase in disequilibrium with the surrounding glass (only in experiment 17-7C). Their composition spans from annite (17-4C) to biotite (17-7C; Supplement Table S1).
The attainment of the equilibrium between amphibole and melt is the fundamental requisite for experimentally deriving the Ds. Due to the complex chemical system, the demonstration of equilibrium through reversal experiments is difficult to achieve. The approach to equilibrium was thus carefully assessed based on the textural relationship between amphibole crystals and glass (Fig. 2), as well as by major and trace element chemical analyses (Figs. 3, 4, and 6). The growth of amphibole crystals occurs at the expense of prior clinopyroxene or as a single crystal crystallized from the melt. In both cases, amphiboles display euhedral habitus, suggesting conditions close to equilibrium with the surrounding glass (Fig. 5). The attainment of equilibrium between amphibole and glass is supported by the relatively homogeneity of major element composition from all experimental runs (Figs. 3 and 4). Further support to the attainment of equilibrium is suggested by consistent trends in the Fe–Mg partitioning between liquids and amphiboles (see Supplement Fig. S2) converging at the ()amph ()glass ratio of ∼ 0.35, except for 18-15HM. The latter is slightly higher (0.42) and is interpreted as being related to a higher amount of Fe loss in the Pt capsule likely due to the different experimental technique with respect to experiment 17-8HM performed at comparable conditions (see above). No significant zoning or heterogeneous REE trace element distribution is apparent in the key sites of the capsule (i.e., top vs. bottom of the capsule; close to vs. far from phase boundaries; Fig. 5). All the above evidence suggests that amphibole and glass are close to equilibrium conditions.
Partition coefficients between amphibole and glass (Ds) reported hereafter are average results for each experimental run and are shown in Fig. 7. Overall, patterns of Ds mostly match with the range defined by the Ds experimentally derived in mafic and intermediate systems (Tiepolo et al., 2007, and references therein; Nandedkar et al., 2016; Fig. 7).
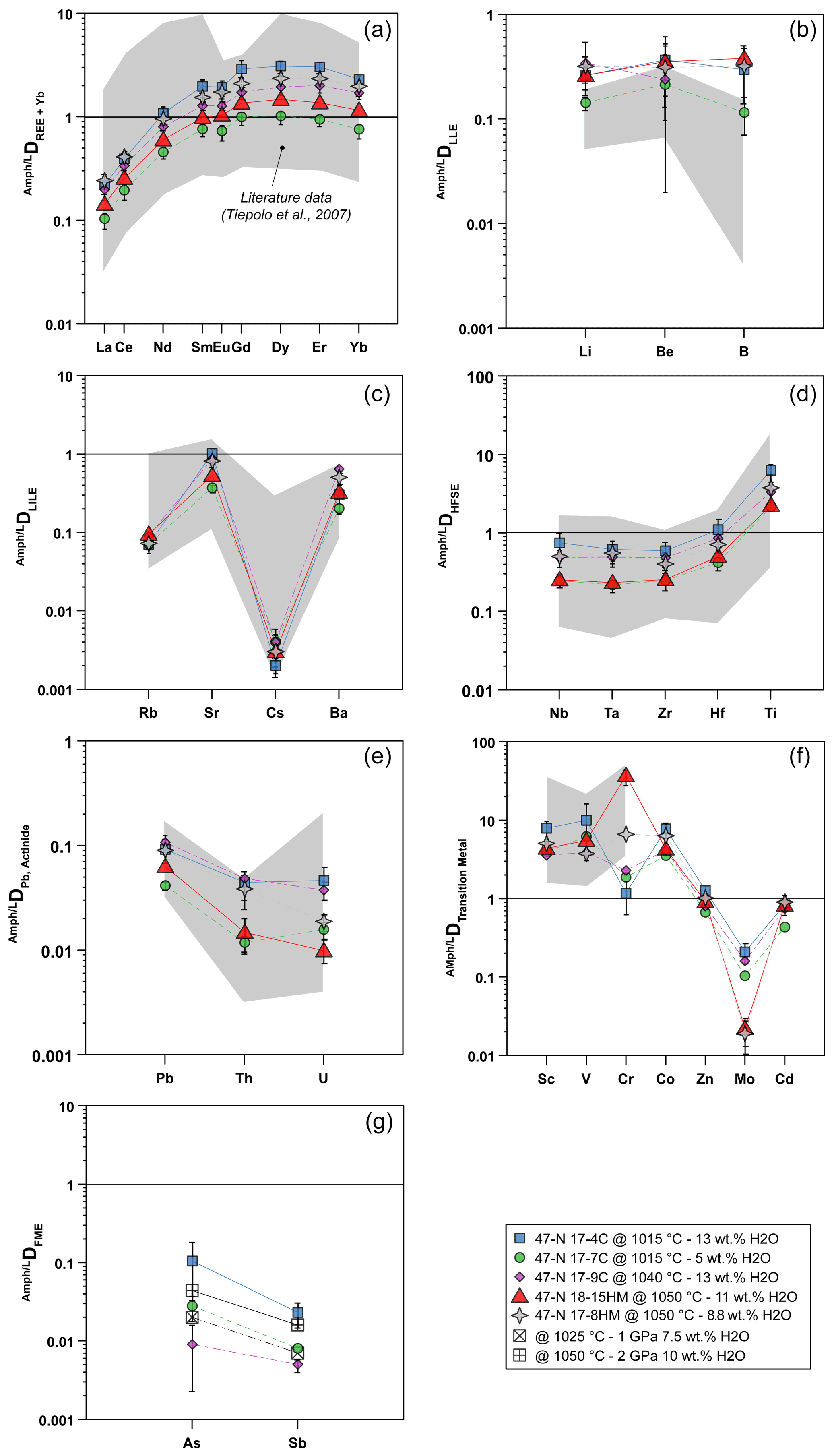
Figure 7Amphibole–liquid partition coefficients (D) for different element groups: (a) REEs, (b) light lithophile elements (LLEs), (c) large ion lithophile elements (LILEs), (d) high field strength elements (HFSEs), (e) Pb and actinide, (f) transition metals, and (g) fluid-mobile elements (FMEs: As, Sb). Gray fields represent a compilation of data from the literature as previously documented by Tiepolo et al. (2007, and references therein) and Nandenkar et al. (2016). Black symbols in (g) refer to D for As and Sb from Adam and Green (2006) performed at different P−T conditions (see legend) and are shown for comparison. When not visible, the error bar is smaller than the symbol.
The DREE patterns are characterized by a depletion in light rare Earth elements (LREEs) relative to M-HREEs (medium and heavy rare Earth elements) that is close to unity in experiment 17-7C and becomes much more compatible in the other experiments in which it reaches values up to 3 (Fig. 7a). The DLa DYb is between 0.09 and 0.14 and reveals a LREE–HREE fractionation slightly more pronounced than that observed in the literature. A slight negative Eu anomaly is appreciable in all the experimental runs, although it is more pronounced in those performed at low fO2 conditions (Fig. 7a).
The light lithophile elements (LLEs – Li, Be, B; Fig. 7b) have similar incompatible behavior in amphibole, and their D varies from 0.114 to 0.375. Remarkably, our results expand to higher values the upper limit of the literature data.
The large ion lithophile elements (LILEs – Rb, Sr, Cs, Ba; Fig. 7c) are moderately (Sr and Ba) to highly (Rb and Cs) incompatible. Cesium is the most incompatible with D between 0.002 and 0.004. The DRb ranges between 0.070 and 0.094, whereas DSr and DBa range from 0.369 to 1.010 and from 0.202 to 0.642, respectively. According to Dalpé and Baker (2000), at comparable T and SiO2 in the coexisting liquid, the DBa of experiments performed at high fO2 is higher compared with those determined in experiments performed at low fO2 (e.g., 17-4C vs. 17-8HM). Partitioning data for LILEs are comparable with those reported in the literature (Fig. 7c).
The high-field strength elements (HFSEs – Nb, Ta, Zr, Hf, Ti; Fig. 7d) are incompatible in amphibole with the exception of Ti that is moderately compatible (DTi=2.1–6.2). The Ds values range from 0.241 to 0.731 for Nb, from 0.212 to 0.608 for Ta, from 0.243 to 0.579 for Zr, and from 0.412 to 1.076 for Hf. The DNb DTa ratio ranges between 0.91 and 1.20 and is in the upper range of ratios reported in the literature (e.g., Tiepolo et al., 2007). Noticeably, new data confirm that in Fe-rich amphiboles (e.g., 17-4C vs. 17-7C) Nb can be more compatible than Ta as already suggested by Tiepolo et al. (2000b). The DZr DHf ratio is in the lower range of the literature data with values from 0.51 to 0.59. Lead and actinides (Th, U) are strongly incompatible, ranging from 0.042 to 0.109 for Pb, 0.012 to 0.091 for Th, and 0.016 to 0.047 for U (Fig. 7e). All values fall within the field of the literature data. The DTh DU is highly variable and spans between 0.734 and 2.086 with the lowest values pertaining to experiments carried out at low fO2.
The D of transition metals (Sc, V, Cr, Co, Zn, Mo, Cd) are reported in Fig. 7f. Scandium, V, Cr, and Co are moderately to highly compatible in amphibole. The D values for Zn and Cd are close to unity and range from 0.672 to 1.276 and from 0.435 to 0.906, respectively. When compared to literature values, D for Sc and V are in the middle of the range variation, whereas DCr is significantly lower for experiments performed at low fO2 (Fig. 7f). The DZn,Cd are close to 1 and comparable with data reported by Adam and Green (2003). Molybdenum is incompatible in all the experiments (from 0.209 to 0.104), especially in those at high fO2 (close to 0.020), and these finding are in agreement with the previous work of Adam and Green (2003) and the high-T experiments of Nanderdkar et al. (2016).
Amphibole–liquid partition coefficients for As and Sb are reported in Fig. 7g. They are moderately to strongly incompatible in experiments at low fO2 and show D values ranging from 0.009 to 0.105 and from 0.005 to 0.023, respectively. The DAs DSb ratio is above unity, decreasing from a maximum of 4.62 for experiment 17-4C (performed at 1015 ∘C) down to 1.76 for experiment 17-9C (performed at 1040 ∘C; Table 5). The available data from the literature are limited to the work of Adam and Green (2006) reporting incompatible behaviors for As and Sb, in agreement with our data at low fO2. Because both As and Sb may form alloys with Pt (see Supplement Fig. S1), the estimation of their Ds at higher fO2 condition is challenging and is dependent on the amount of As and Sb escaping from the system during the experimental runs. Considering that As and Sb concentrations of the glass in equilibrium with the amphibole are very low (below 5 and 3 ppm, respectively; see Table 3) compared to their concentrations in the starting materials (Table 1), we conclude that the formation of As–Sb alloy with the Pt capsule during the experimental run may have subtracted these elements from the silicate melt and in turn increased their partition coefficients. Therefore, the As and Sb concentrations of both amphibole and glass from experiments performed at high fO2 conditions cannot be used to constrain their Ds, and further experimental work is required.
Table 5Amphibole–glass partition coefficients calculated from LA-ICP-MS analyses of run products.

* SD – standard deviation. nd – not determined. D in italics (As and Sb in HM experiments) are not considered reliable (see text).
Chlorine data from EMPA were used to calculate the D of Cl, which results in a variation between 0.24 and 0.44 (Table 5). No apparent relation with the melt composition nor with the fO2 conditions of the experimental run is appreciable. The obtained data confirm the incompatibility of Cl in Ca-amphibole, as reported in previous works (Dalou et al., 2014; Van Den Bleaker and Koga, 2015; Bénard et al., 2017), with the new DCl falling at the upper limit of the literature data (ranging from 0.03 to 0.62; median at 0.26).
Trace element partitioning between minerals and liquid is a complex function of the phase compositions, as well as the intensive parameters of the system (i.e., P−T conditions). Melt polymerization has been shown to strongly influence Ds (Tiepolo et al., 2000a). In addition, the crystal structure filters cations based on the affinity in charge and ionic radius with the major cations of each crystal site. The average dimension of the structural site, which can be considered equivalent to the ideal radius (r0) parameter of Blundy and Wood (1994), rules the uptake of trace elements with the same valence. Variations in the major element site occupancy can induce modifications of r0 and, in turn, the relative compatibility of trace elements with a different ionic radius entering the same site (Tiepolo et al., 2000a). Modifications of the formal charge of the site have consequences for the compatibility of cations favoring the isovalent substitutions (Blundy and Wood, 1994).
Table 6Lattice parameters calculated from partition coefficient data for the M(1-3) site in amphibole for experiments with comparable SiO2 but different fO2.
6.1 The effect of melt composition on Ds
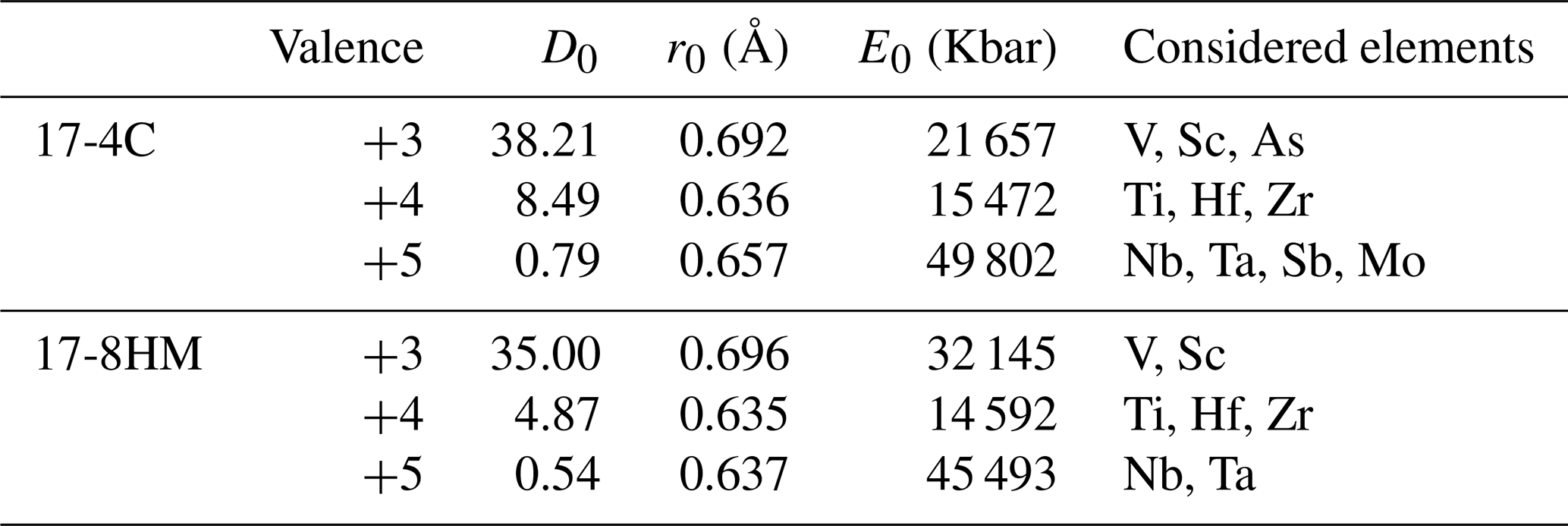
Because chemistry and polymerization of melt are parameters that mostly influence the Ds (e.g., Tiepolo et al., 2007, and references therein), the low-charge trace elements with a relatively large ionic radius (e.g., Rb1+) have affinity with network-forming cations and their compatibility with amphibole is expected to decrease as the melt becomes SiO2- and alkali-rich. The opposite behavior is observed for trace elements with strong network modifier behavior (low ionic radius and high charge, e.g., Nb5+).
The compositional variations in the melt in equilibrium with the experimental amphibole of this study are rather narrow. The SiO2 content of the glass spans from 52.3 wt % to 57.9 wt %, and Al2O3 and alkali abundances show limited variations (Table 3; Fig. 3a). However, we observed the most significant difference in the trace element partitioning as a function of SiO2 content of equilibrium melt. The D increases up to 3 times with the increasing of the SiO2 content in the melt for those elements with an affinity for network-modifying cations such as Sr and HFSEs (Fig. 8a), as well as Y and REEs (Fig. 8b). A decrease in compatibility is reported for those elements having affinity with the network forming cations such as Rb and Cs (Fig. 8c). The influence of melt composition on the D is not easily evaluable for elements with multiple valence (see next section). However, if we distinguish low and high fO2 experiments, the dependence of D on melt composition is more evident. This is the case, for example, of Mo and U that at low fO2 increase their compatibility for amphibole as the SiO2 content in the melt increases. At high fO2, given the extremely high incompatibility of these elements (particularly for Mo), the increase in D at the increasing SiO2 is less evident (Fig. 8d).
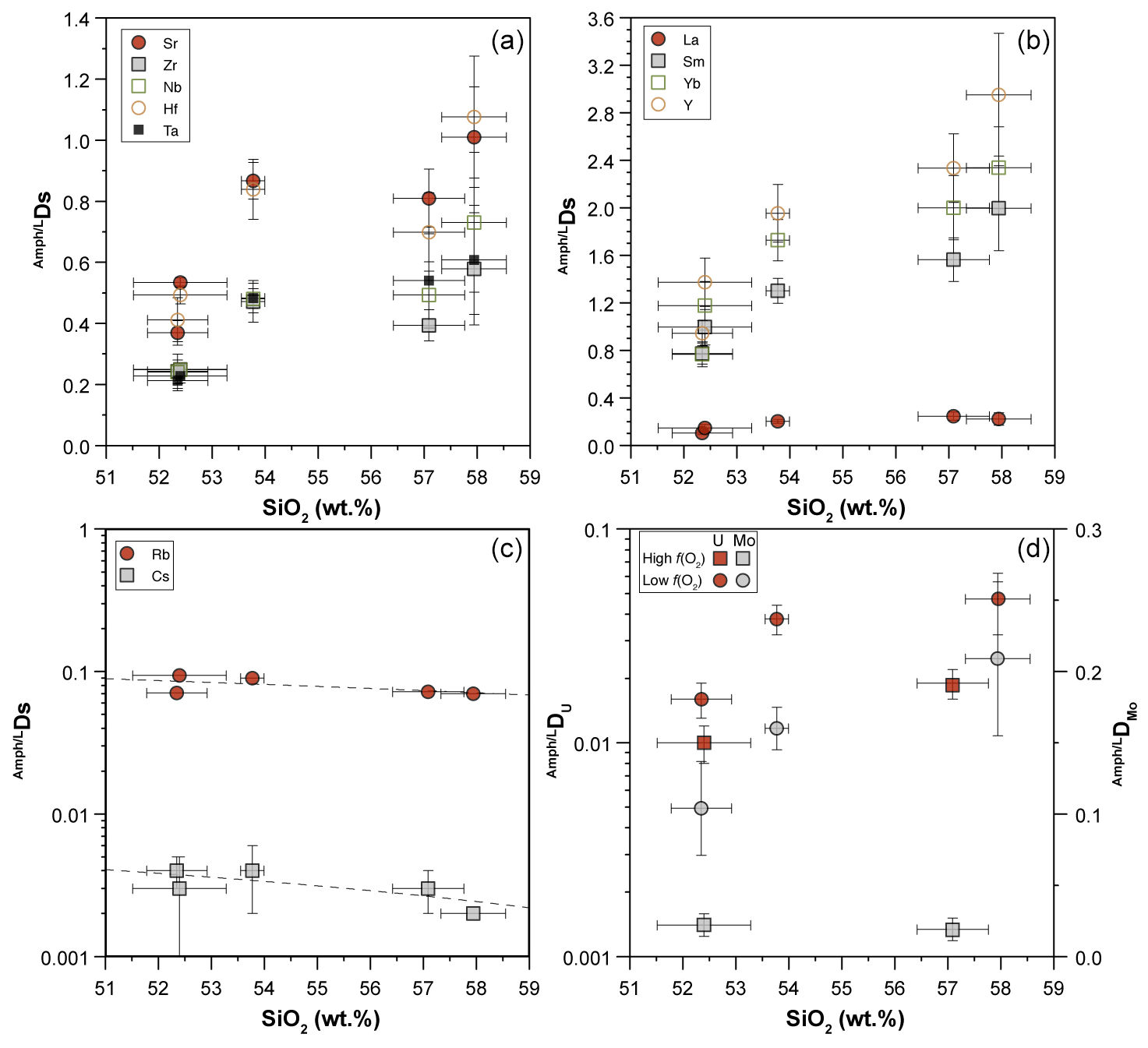
Figure 8Correlations between the SiO2 content (wt %) of the liquid in equilibrium with the amphiboles and the D of key elements: Sr and HFSEs in (a), REEs + Y in (b), Rb and Cs (LILEs) in (c), and elements with multiple oxidation state (U and Mo) in (d).
6.2 The effect of fO2 on Ds
The fO2 variation may affect trace element partitioning between minerals and melt in different ways. First, fO2 may induce variations in the oxidation state of a trace element, thus modifying the crystal chemical mechanism responsible for its incorporation. Secondly, it may affect the oxidation state of a major elements (e.g., Fe). This leads to a variation in the formal charge of the crystal site with implications in the capability to host a given trace element.
In order to isolate, as much as possible, the effect of fO2 variation, we compared in Fig. 9 the Ds for amphiboles, distinguishing those equilibrated with melt at low and high SiO2 and different fO2 conditions (see also Fig. 8d). A common feature is that the D of Ni, Ti, V, Mo, and U vary significantly within the range of investigated fO2. At nearly constant melt composition, the compatibility of Ti, V, Mo, and U in amphibole decreases under high fO2, whereas that of Ni is enhanced (Fig. 9).
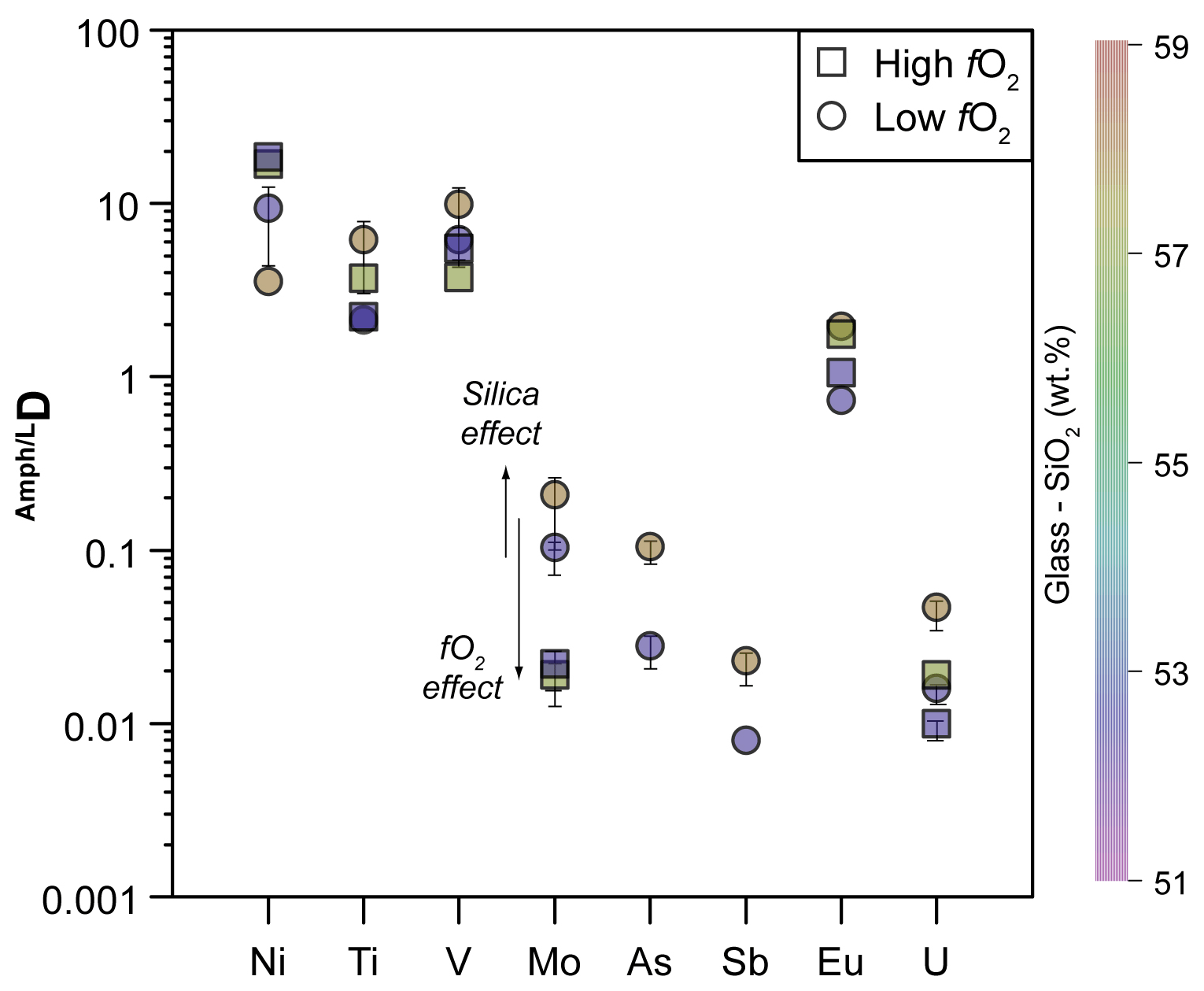
Figure 9Comparison between the D of trace elements with multiple oxidation states obtained at low (circles) and high (squares) fO2 conditions and similar SiO2 content (wt %) of the glass in equilibrium with the amphiboles (color code on the right side of the diagram). Arrows show general trends induced by both silica content in the glass and fO2 variations. See text for further explanation. When not visible, the error bar is smaller than the symbol.
In order to describe the incorporation of trace elements in the amphibole structure, we follow the lattice-strain model of Blundy and Wood (1994). Partition coefficients of elements with the same charge and ionic radius entering in the same crystal site lie on parabolas described by an equation with three variables – D0, r0, and E – that are the partition coefficient, the ionic radius of the ideal cation of the crystallographic site (which does not require work for substitution), and the Young's modulus of the site, respectively. We applied the calculation to amphiboles from experiments 17-4C and 17-8HM for which the glasses in equilibrium with amphibole show comparable SiO2 contents (Table 3). The results for the M(1-3) site are reported in Table 6 and in Fig. 10. Ideal r0, D0, and E were calculated by least-squares fitting of the measured partition coefficients to the theoretical curve given by the equation of Blundy and Wood (1994). Although trace elements are generally incorporated and distributed among the three octahedral sites of amphiboles M(1-2-3), no attempt was made to either take account for or model the split site behavior (i.e., M(1) vs. M(2) vs. M(3)).
Figure 10Measured partition coefficients and fitted solution to the lattice-strain model for the M(1–3) site (i.e., Onuma diagrams) in amphibole of experiments 17-4C (a) and 17-8HM (b) as representative of low and high fO2 conditions, respectively (data in Table 6). Ionic radius data (in Å) from Shannon (1976). To note is that the parabola for 5+ cations for experiment 17-8HM assumes that Young's modulus does not change significantly with fO2, as suggested by the similarity in the Young's modulus for 4+ cations that are more constrained for experiments at both fO2 conditions (see Table 6).
Titanium is assumed to be 4+ and is not expected to vary the oxidation state under the investigated range of fO2. Variations in the D from high to low fO2 conditions (Fig. 9) are thus not directly related to the Ti oxidation state but likely to changes in the crystal chemical mechanism of its charge balance. High-T amphiboles are commonly characterized by an oxo-component at the O(3) site that, according to Oberti et al. (2007) and Popp et al. (1995, 2006), can be balanced by the following crystal chemical mechanisms involving either Ti4+ or Fe3+:
High Fe3+ Fetot ratios will favor the second crystal chemical mechanism with respect to that involving Ti in the M(1) site with the consequence of a lower compatibility of Ti in the amphibole structure. An accurate estimate of the Fe3+ Fetot in the system is not straightforward. In particular, given the large number of variables in the amphibole structure such as the effective water content or the A site occupancy, the Fe3+ Fetot ratio cannot be inferred from the stoichiometry. However, amphiboles synthetized at higher fO2 possess, on average, higher Fe3+ Fetot (0.161 in experiment 18-15HM). We thus suggest that the decrease in the Ti compatibility at high fO2 conditions is likely related to a competition with Fe3+ in the balance of the oxo-component with the consequence that mechanism 2 is thus preferred in high-T amphiboles. Noticeably, the decrease in Ti compatibility is coupled with a decrease in the D of other high charge cations such as Zr–Hf and Nb–Ta, which substitute for Ti in the amphibole structure (Tiepolo et al., 2011). This is in agreement with the evidence that Nb and Ta contribute in the balance of the oxo-component (Tiepolo et al., 2000b). We cannot exclude, however, that the higher formal charge of the octahedral sites related to the higher proportion of Fe3+ hampers the incorporation of other high charge cations.
Many transition metals (V, Co, Ni, and Mo) are characterized by multiple oxidation states, and, thus, the changes in fO2 conditions induced in the system may have influenced their partitioning. They are incorporated and distributed among the three octahedral sites of amphiboles M(1-2-3); for this reason, robust conclusions on their oxidation state at the investigated conditions is not straightforward. Results show that no significant difference between DV and DCo at low and high fO2 conditions occurs (Fig. 7f). Vanadium compatibility is comparable to that of Sc3+ (Fig. 7f), and, thus, we suggest that V3+ is the dominant form in all experiments (Fig. 10). However, a slight decrease in the DV in the experiments performed at higher fO2 conditions is observed (Fig. 9). Our experiments cannot constrain the oxidation state of Co. The similarity of the obtained DCo at low fO2 with those determined by Adam and Green (2006) at comparable conditions suggests that Co2+ is dominant. The absence of a significant change in Co compatibility with the increased fO2 (Fig. 7f) indicates that the transition from 2+ to 3+ has not occurred. According to Dingwell et al. (1994), Ni2+ should be dominant in the range of log fO2 between −13.5 and −8.5. Our experiments at low fO2 conditions are within this range (Fig. 1c), and we can, thus, reasonably assume that in amphibole Ni2+ is dominant. In the experiments performed at higher fO2 (up to log fO.5; Fig. 1c), Ni compatibility in amphibole significantly increases (up to 5 times; Fig. 9). Because all other conditions were kept almost constant, the higher compatibility of Ni2+ is difficult to explain without a change in the oxidation state. Therefore, we suggest that at the higherfO2 conditions Ni2+ is partially oxidized to Ni3+ whose ionic radius is very close to that of Fe3+ ([6]0.60 and [6]0.645 Å, respectively; against [6]0.69 Å for Ni2+; Shannon, 1979). Molybdenum has multiple oxidation states, and, according to Adam and Green (2006), it can be incorporated as a pentavalent cation in the amphibole's structure at low fO2 conditions. The decrease in its compatibility with the increase in fO2 suggests that under more oxidized conditions Mo5+ is converted to Mo6+. Due to the high charge and the lack of suitable charge balance mechanisms, its compatibility for amphibole is strongly reduced (Figs. 8d, 10).
Arsenic and Sb both have 3+ and 5+ oxidation states. In the experiments at low fO2, As is highly incompatible, and its D can be fitted in terms of the elastic strain model with that of V3+ and Sc3+ (Fig. 10a). The resulting r0 of the site is similar to that of the curve for the 4+ cations (Table 6) and in agreement with the dimensions of an octahedral site of amphibole (e.g., Tiepolo et al., 2000a). Although As, V, and Sc can be fitted with a single parable, the simulation of their behavior is not fully reliable. The reader would consider that these elements may be distributed among the different octahedral sites – M(1), M(2), M(3) – and, thus, the fit reported in Fig. 10 could represent an average. Furthermore, we cannot exclude that both As3+ and As5+ are hosted in the amphibole's structure. However, we can conclude that the prevalent oxidation state of As at the low fO2 conditions is 3+. The DSb at low fO2 conditions is very low, even lower than that of the 5+ cations Nb, Ta, and Mo. If the DSb at low fO2 conditions is fitted with Nb5+, Ta5+, and Mo5+ (Fig. 10a), a site with a r0 very close to that of the 4+ cations is obtained. This would suggest that an oxidation state of 5+ under the investigated conditions is likely. At comparable fO2 conditions, Adam and Green (2006) found that in amphibole crystallized at 2.0 GPa and 1050 ∘C Sb also has a 5+ oxidation state of Sb. Although the DAs and DSb at high fO2 conditions are not available from our experimental investigations, we estimate that DSb should increase up to 0.113, according to the fitted solution to the lattice-strain model reported in Fig. 10b for 5+ cations for the M(1–3) site. The increase in the DSb at higher fO2 conditions cannot be explained by changes in the oxidation state and could be reasonably related to the presence of the oxo-component requiring Fe3+ in the M(1) site. We propose that the similarity in the ionic radius between Sb5+ and Fe3+ (0.60 and 0.645 Å, respectively; Fig. 10b) is responsible for the increase in the DSb. On the contrary, the D for As3+ is expected to decrease its compatibility down to a value as low as 0.004. However, an increase in the DAs will occur considering a lower Young's modulus of the M(2) site (i.e., less narrow parabola of the 3+ cations). Generally, the 3+ cations enter the M(2) site of amphibole (Oberti et al., 2007); however, Fe3+ is required also in the M(1) site for charge balance in the presence of the oxo-component. Given the affinity in the ionic radius between As3+ and Fe3+, we expect that both cations are partitioned between M(1) and M(2) and, thus, likely decoupled from V and Sc with concurrent potential increase in the DAs. Change in the oxidation state of As from 3+ to 5+ due to higher fO2 conditions is expected to decrease the D because [6]As5+ has ionic radii of 0.46 Å (Shannon, 1976), which is too small for the M(1–3) site (Fig. 10b). The incorporation of [4]As5+ (ionic radii of 0.335 Å) is unlikely.
Uranium can be either 4+ or 6+, whereas Th is only 4+. Except for run 17-9C (Cl-rich experiment, see next section), experiments at low fO2 conditions result in DTh DU ratios lower than unity, whereas experiments at higher fO2 conditions show greater than unity ratios. We interpret this feature as related to the change in the fO2 of the system and, in particular, to the partial oxidation of U from 4+ to 6+. At fO2 around 2 log units below the FMQ buffer, a condition close to those of our experiments with the graphite sleeve (Fig. 1c, Table 2), U and Th are expected to enter the amphibole structure as 4+ (Tiepolo et al., 2000a) and are incorporated within the M(4) site. At high fO2 conditions U is likely partially oxidized to 6+, which makes the substitution with Ca at the M(4) site more difficult and causes a drastic reduction of its partition coefficient. Remarkably, the occurrence of U6+ at fO2 conditions above the FMQ buffer is reported in previous experimental work (e.g., Fonseca et al., 2014), thus supporting the proposed scenario. A complete oxidation of U to 6+ in our experiments is unlikely because an even higher incompatibility would be expected.
6.3 The effect of Cl on Ds
Amphiboles in this experimental study are characterized by variable Cl contents that may reach up to 0.88 wt % (Table 4). Despite the relatively high Cl content in our amphiboles, it behaves as a moderate incompatible element, as already reported in the literature (Dalou et al., 2014; Van Den Bleaker and Koga, 2015; Bénard et al., 2017). However, the incorporation of a higher amount of Cl into Ca-amphibole at the O(3) site may significantly modify its crystal structure (Hawthorne and Oberti, 2007; Oberti et al., 2007, 1993). The larger ionic radius of Cl ([6]Cl.81 Å; Shannon, 1976) with respect to substituting anions ([6]O.40 Å, [6]OH.37 Å, [6]F.33 Å) causes an increase in the anionic site size with a consequent modification of the <M(1)-O(3)> and <M(3)-O(3)> distances. Therefore, the expansion of the octahedral sheet in response to Cl incorporation requires a concomitant expansion of the tetrahedral chain and a reduction of the A site (Oberti et al., 1993). All these interlocked structural changes induced by Cl incorporation affect the trace element incorporation in amphibole.
Because the entrance of Cl in the amphibole structure does not cause charge disequilibria but only variations in the site dimensions, its effect is mainly on the ratios between the Ds. A clear effect of the entrance of Cl in the amphibole structure is on Th and U partitioning. Two positive trends, pertaining to low and high fO2 conditions, between DTh DU ratio and Cl content in amphibole can be identified albeit a relatively small number of observations and their uncertainties (Fig. 11a). Tetravalent Th has a ionic radius larger than U4+ ([8]1.05 vs. [8]1.00 Å, respectively), and their incorporation should occur predominantly at the M(4) site, as previously discussed. The observed increase in the compatibility of Th relative to that of U is likely related to an expansion of the M(4) site in relation to the increased dimension of the O(3) site due to the presence of Cl. The results of such a mechanism is depicted in Fig. 12 where the calculated r0 of 4+ cations entering in the M(4) site increase, together with the increasing of the amphibole's Cl content. In a similar way, the increasing of the Cl content in amphibole negatively correlates with the DGd DYb ratios (Fig. 11b), elements that should be incorporated in the M(4) site. However, this trend shows opposite behavior compared to the DTh DU ratio (Fig. 11a), suggesting the preferential incorporation of smaller cations (e.g., Yb) over the larger ones (e.g., Gd). This behavior suggests a decrease in the M(4) site in response to the Cl incorporation within amphibole, which appears in contrast to what is proposed by Oberti et al. (1993) and the finding reported in this study (Fig. 12). A possible explanation for such behavior is a potential r0 decoupling between the 4+ and 3+ cations in the same site (e.g., Shimizu et al., 2017) or, alternatively, the distortion of the M(4) site induced by Cl incorporation in the O(3) anionic site that may disturb the ordering of the REE3+ in the M(4) site, splitting them and enhancing the incorporation of HREEs in the smaller M(4′) site. The latter scenario resemble the “cummingtonite component” often characterizing the high-T kaersutite as discussed by Bottazzi et al. (1999).
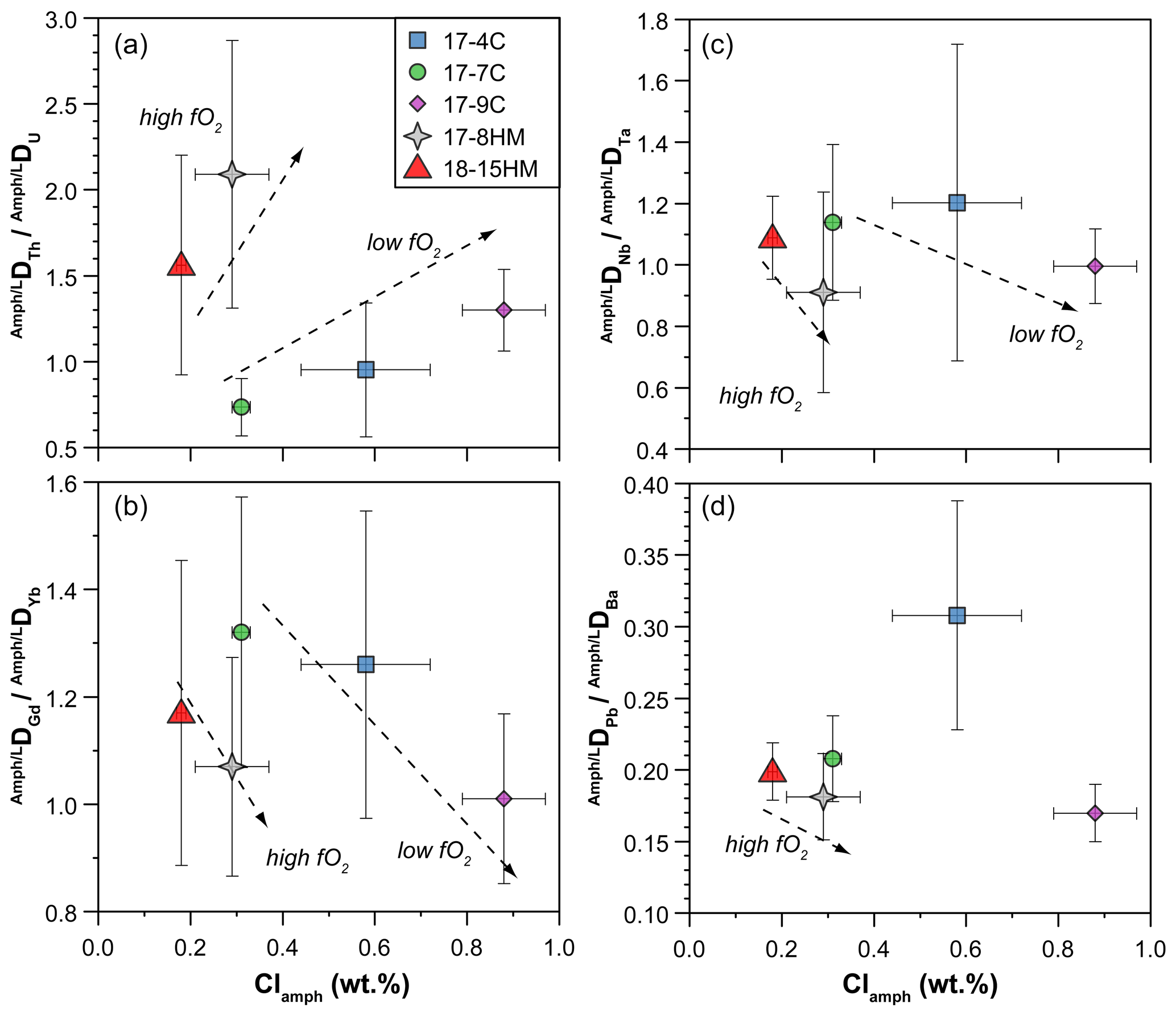
Figure 11Chlorine content (wt %) in amphibole vs. DDU (a), DDYb (b), DDTa (c), and DDBa (d) ratios measured in the experiments. See text for details.
Our data also evidence a correlation between the amphibole Cl content and the DNb DTa ratios (Fig. 11c, d), trace elements usually incorporated within the M(1) site of amphiboles. According to Tiepolo et al. (2000b), Nb should be slightly larger than that of Ta (about 0.65 and 0.64 Å, respectively), and, because the DNb DTa ratio is related to the <M(1)-O> distance in Ti-rich pargasite and kaersutite (Tiepolo et al., 2000b), the DNb DTa ratio is expected to increase due to the variation in this distance related to O(3)Cl incorporation. However, our experiments show a negative correlation between the Cl content in amphibole and the DNb DTa ratio at both low and high fO2 conditions (Fig. 11c), suggesting that the M(1) octahedron decreases at the diminishment of the <M(1)-O(3)> distance. The application of the relationship between the Mg# and Ti content (a.p.f.u.) of amphibole and the <M(1)-O> distance provided by Tiepolo et al. (2000b) allows us to calculate the <M(1)-O> distance of amphiboles in our experiments. These calculations support a decreasing of the <M(1)-O> distance coupled with the increasing of the Cl content of our amphibole, varying from 2.081 to 2.077 Å for Cl-poor (17-7C) and Cl-rich (17-9C) experiments, respectively. According to Tiepolo et al. (2007), high Ti content in Ca-amphibole can induce Ti to occupy the M(1) site slightly shifted along the b crystallographic axis of amphibole towards the O(3)-O(3) edge, named M(1′). This local coordination is smaller than that of the dominant cations (mainly Mg and Fe2+) and may account for the higher incorporation of Ta over Nb, thus explaining the observed trend.
Figure 11d displays the negative correlation between the Cl content in amphibole and DPb DBa ratio in low and high fO2 experiments. These bivalent elements, among which Ba is slightly larger than Pb (rBa=1.61 and rPb=1.49 Å, respectively; Shannon, 1976), are both incorporated within the A site of the amphibole, and the ratio of their D is expected to be strictly related to the A site dimensions. The larger increase in the DBa compared to that of DPb is thus accounted for by the increase in the A size dimensions in response to the higher Cl fraction in amphibole. However, this finding appears to be in contrast with the observation of Oberti et al. (1993) who individuate a reduction of the A site as a result of tetrahedral chain expansion due to Cl incorporation.
Further modification of Ds due to the incorporation of Cl in the O(3) site has been recently documented for the OH−. The effect of Cl content in amphibole on its water incorporation has been investigated by using a novel statistical and methodological approach based on a micro-Raman technique (Cannaò et al., 2022). In a separate paper, we measured the H2O content in both amphibole and glass in the experiments presented in this study to calculate the partition coefficient of water (D). These new D data indicate that high Cl content in amphibole may increase its water affinity, thus suggesting that crystallographic features may also control the hydrogenation of amphibole.
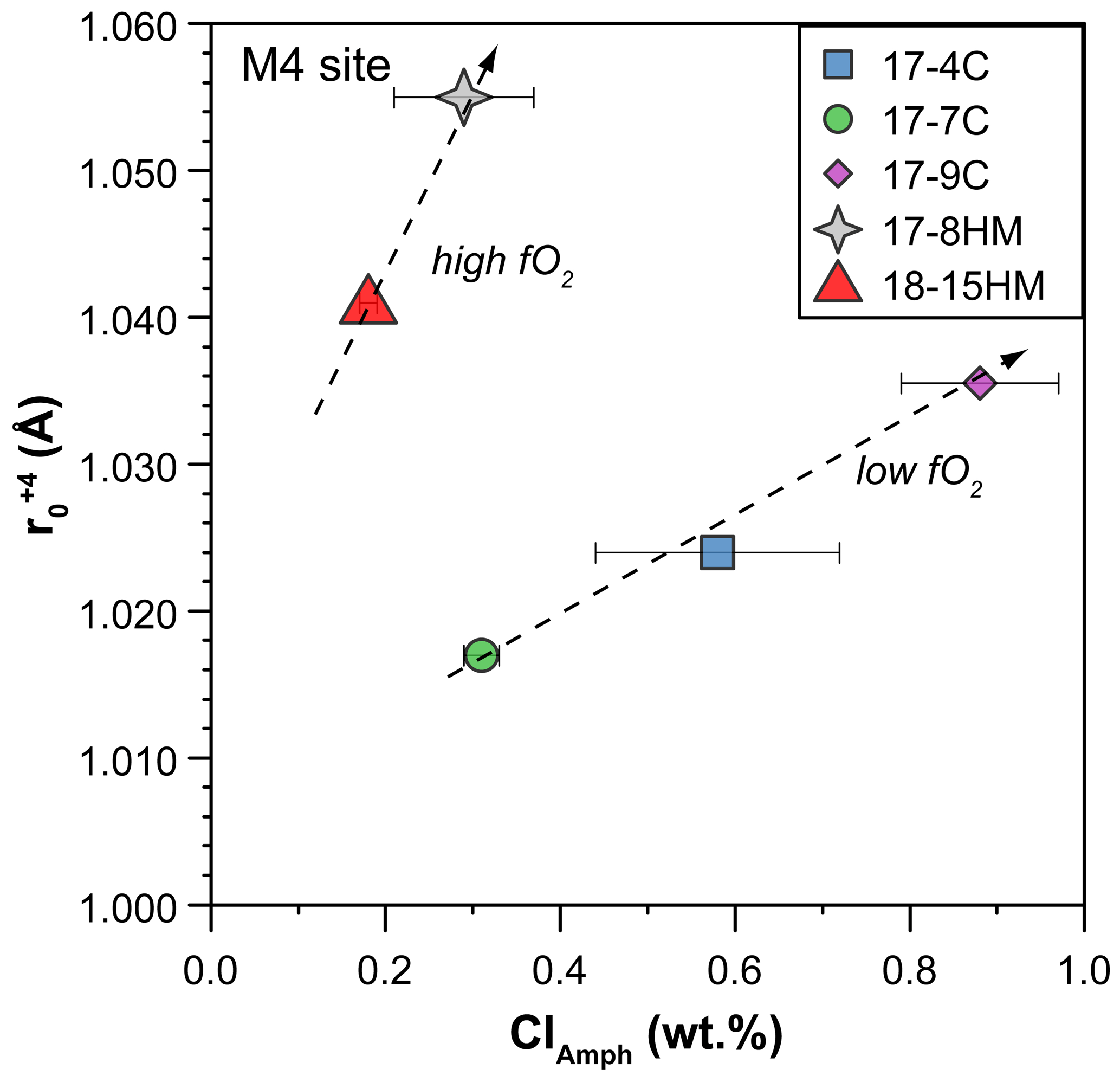
Figure 12Relationship between Cl content in amphibole (in wt %) and the ideal radius (r0 in Å) of the M(4) site for +4 cations as calculated by using the measured partition coefficients of U and Th and fitted solution to the strain-lattice model of Blundy and Wood (1994). Increase in r0 of the M(4) site due to Cl enrichment in amphibole allows Th4+ (rTh=1.05 Å) to be more compatible than U4+ (rU=1.00 Å). Data are reported in Table S3 of the Supplement, and the complete Onuma diagrams of the M(4) site are illustrated in Fig. S3 of the Supplement.
In this experimental work we provided new Ds for a large number of trace elements including some with still uncertain behavior (e.g., As, Sb, and Mo). For the latter, available data are currently limited to a few works dealing with peculiar system compositions (e.g., komatiites; Fiorentini et al., 2012). Moreover, data on high chlorine amphiboles (up to 0.47 wt %) are scarce and their trace element compositions completely unknown (source http://georoc.mpch-mainz.gwdg.de, last access: 5 December 2021). Below, we provide several general implications based on the main results of our work.
The alkali-rich (Na2O + K2O between 6.0 wt % and 7.7 wt %) and intermediate silica compositions (from 52.3 wt % to 57.9 wt %) of the liquid coexisting with the kaersutitic amphibole in our experiments allow us to apply the obtained partitioning data to several geological settings and, particularly, those characterized by a volatile-rich and variable oxidized upper-mantle environment. Although alkaline magmas are predominantly reported in continental regions, they can also occur in oceanic and subduction zone settings (Davies et al., 1989; Lange and Carmichael, 1991). There is a general consensus in considering alkaline magmas as the result of a low degree of partial melting of volatile-rich mantle sources metasomatised by fluids or melts derived from previously subducted slabs (Hofmann, 1997; Menzies and Murthy, 1980; Pilet et al., 2008). More recently, it has been proposed that, in subduction settings, alkaline magmas can also be generated by the partial melting of mélange diapirs at the base of the lithosphere (Cruz-Uribe et al., 2018). In all cases, the influence of slab-derived materials significantly impacts on the trace element and volatiles budget of the produced melts, which will be enriched in Cl and other fluid-mobile elements, such as As, Sb, Mo, Th, U, and Pb (e.g., Bali et al., 2012, 2011; Barnes et al., 2018; Scambelluri et al., 2019, and references therein). A recent experimental investigation remarks how the presence of high-Cl content in slab-derived fluids enhances the mobility of trace elements such as LILEs, LREEs, Pb, and U (Rustioni et al., 2019).
Based on the results of our study, the capability of amphibole to act as filter for trace elements at upper-mantle levels is influenced by the redox condition of the system and of the Cl content of the amphibole itself. In particular, at relatively high fO2 conditions both U and Mo significantly increase their incompatibility. The increase in the fO2 and Cl content of a system allows U and Mo to be extensively extracted from an amphibole-bearing mantle source and transferred to upper-crustal levels via melts.
We also have experimentally demonstrated that the DV decreases at increasing fO2 (Fig. 9). This results in a general decrease in the DV Amph/LDSc ratio, which is consistent with the trends reported for the same partition coefficients for olivine, pyroxenes, and spinel (Lee et al., 2005). This evidence may be of particular interest and represents a potential tool to estimate the fO2 conditions during amphibole crystallization, although more experimental investigations are required to cover a wider range of fO2 conditions. Remarkably, our interpretation of the DV DSc ratio variation does not concern the direct oxidation of V from 3+ to 4+ (or 5+) but rather is related to the conversion of Fe2+ to Fe3+, for which V is more similar in terms of ionic radius dimension ([6]Fe.780 Å vs. [6]Fe.645 Å; [6]V.640 Å vs. [6]V.54 Å; [6]Sc.745 Å; Shannon, 1976).
The distortion of the amphibole crystal structure due to the incorporation of Cl in the O(3) site can lead to a relative Th enrichment with respect to U and Pb concentrations in amphibole (Fig. 11a, d). Although Pb is generally more compatible than actinides (Tiepolo et al., 2000a), the relatively higher Th incorporation may impose a decoupling behavior between Th and U–Pb potentially affecting the Pb isotope systematic over long geological timescales. In particular, we speculate that such decoupling potentially will produce amphibole with more radiogenic 208Pb 204Pb ratio than the Pb-daughter isotopes decaying from the U-series (i.e., 207Pb and 206Pb).
All data derived from this research are presented in the enclosed tables, figures, and Supplement files.
The supplement related to this article is available online at: https://doi.org/10.5194/ejm-34-35-2022-supplement.
EC and MT conceptualized the project. EC performed the experiments with the supervision of GB and PF. EC carried out the EMPA characterization and the LA-ICP-MS analyses with the guidance of MT and AL. All authors discussed and interpreted the results. Acquisition of the financial support for the project was provided by PF. The manuscript was written by EC with contributions from all co-authors.
The contact author has declared that neither they nor their co-authors have any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue “Probing the Earth: experiments and mineral physics at mantle depths”. It is not associated with a conference.
This work was supported by the Italian Ministry of Education, University and Research (MIUR), project “Programma di Rilevante Interesse Nazionale” – (PRIN 2015C5LN35) “Melt-rock reaction and melt migration in the MORB mantle through combined natural and experimental studies”. Constructive reviews by Stephen Foley and an anonymous reviewer, as well as manuscript handling by Didier Laporte (associated editor) and Elisabetta Rampone (chief editor), have been much appreciated.
This research has been supported by the Ministero dell'Istruzione, dell'Università e della Ricerca (grant no. PRIN2015C5LN35).
This paper was edited by Elisabetta Rampone and reviewed by Stephen Foley and one anonymous referee.
Adam, J. and Green, T.: The influence of pressure, mineral composition and water on trace element partitioning between clinopyroxene, amphibole and basanitic melts, Eur. J. Mineral., 15, 831–841, https://doi.org/10.1127/0935-1221/2003/0015-0831, 2003.
Adam, J. and Green, T.: Trace element partitioning between mica- and amphibole-bearing garnet lherzolite and hydrous basanitic melt: 1. Experimental results and the investigation of controls on partitioning behaviour, Contrib. Mineral. Petr., 152, 1–17, https://doi.org/10.1007/s00410-006-0085-4, 2006.
Bali, E., Audétat, A., and Keppler, H.: The mobility of U and Th in subduction zone fluids: An indicator of oxygen fugacity and fluid salinity, Contrib. Mineral. Petr., 161, 597–613, https://doi.org/10.1007/s00410-010-0552-9, 2011.
Bali, E., Keppler, H., and Audetat, A.: The mobility of W and Mo in subduction zone fluids and the Mo-W-Th-U systematics of island arc magmas, Earth Planet. Sc. Lett., 351–352, 195–207, https://doi.org/10.1016/j.epsl.2012.07.032, 2012.
Barnes, J. D., Manning, C. E., Scambelluri, M., and Selverstone, J.: The Behavior of Halogens During Subduction-Zone Processes, in: The role of Halogens in terrestrial and extraterrestrial geochemical processes, edited by: Harlov, D. and Aranovich, L., Springer, Berlin, Germany, 46 pp., https://doi.org/10.1007/978-3-319-61667-4_8, 2018.
Bénard, A., Koga, K. T., Shimizu, N., Kendrick, M. A., Ionov, D. A., Nebel, O., and Arculus, R. J.: Chlorine and fluorine partition coefficients and abundances in sub-arc mantle xenoliths (Kamchatka, Russia): Implications for melt generation and volatile recycling processes in subduction zones, Geochim. Cosmochim. Ac., 199, 324–350, 2017.
Blundy, J. and Wood, B.: Prediction of crystal-melt partition coefficients from elastic moduli, Nature, 372, 452–454, https://doi.org/10.1038/372452a0, 1994.
Bottazzi, P., Tiepolo, M., Vannucci, R., Zanetti, A., Brumm, R., Foley, S. F., and Oberti, R.: Distinct site preferences for heavy and light REE in amphibole and the prediction of Amph/LD REE, Contrib. Mineral. Petr., 137, 36–45, 1999.
Campanaro, B. P. and Jenkins, D. M.: An experimental study of chlorine incorporation in amphibole synthesized along the pargasite-ferro-pargasite join, Can. Mineral., 55, 419–436, https://doi.org/10.3749/canmin.1600082, 2017.
Cannaò, E. and Malaspina, N.: From oceanic to continental subduction: Implications for the geochemical and redox evolution of the supra-subduction mantle, Geosphere, 14, 2311–2336, https://doi.org/10.1130/GES01597.1, 2018.
Cannaò, E., Schiavi, F., Casiraghi, G., Tiepolo, M., and Fumagalli, P.: Effect of chlorine on water incorporation in magmatic amphibole: experimental constraints with a micro-Raman spectroscopy approach, Eur. J. Mineral., 34, 19–34, https://doi.org/10.5194/ejm-34-19-2022, 2022.
Chan, A., Jenkins, D. M., and Dyar, D. M.: Partitioning of chlorine between NaCl brines and ferro-pargasite: Implications for the formation of chlorine-rich amphiboles in mafic rocks, Can. Mineral., 54, 337–351, https://doi.org/10.3749/canmin.1500043, 2016.
Chou, I. M., Eugster, H. P., Berens, P., and Weare, J. H.: Diffusion of hydrogen through platinum membranes at high pressures and temperatures, Geochim. Cosmochim. Ac., 42, 281–288, https://doi.org/10.1016/0016-7037(78)90181-3, 1978.
Coltorti, M., Beccaluva, L., Bonadiman, C., Faccini, B., Ntaflos, T., and Siena, F.: Amphibole genesis via metasomatic reaction with clinopyroxene in mantle xenoliths from Victoria Land, Antarctica, Lithos, 75, 115–139, https://doi.org/10.1016/j.lithos.2003.12.021, 2004.
Connolly, J. A. D.: Computation of phase equilibria by linear programming: A tool for geodynamic modeling and its application to subduction zone decarbonation, Earth Planet. Sc. Lett., 236, 524–541, https://doi.org/10.1016/j.epsl.2005.04.033, 2005.
Cruz-Uribe, A. M., Marschall, H., Gaetani, G. A., and Le Roux, V.: Generation of alkaline magmas in subduction zones by melting of mélange diapirs, Geology, 46, 2–5, https://doi.org/10.1130/G39956.1, 2018.
Dalou, C., Koga, K. T., Le Voyer, M., and Shimizu, N.: Contrasting partition behavior of F and Cl during hydrous mantle melting: implications for Cl/F signature in arc magmas, Prog. Earth Planet. Sci., 1, 26, https://doi.org/10.1186/s40645-014-0026-1, 2014.
Dalpé, C. and Baker, D. R.: Experimental investigation of large-ion-lithophile-element-, between calcic amphibole and basaltic melt: the effects of pressure and oxygen fugacity, Contrib. Mineral. Petr., 140, 233–250, 2000.
Davidson, J., Turner, S., Handley, H., Macpherson, C., and Dosseto, A.: Amphibole “sponge” in arc crust?, Geology, 35, 787–790, https://doi.org/10.1130/G23637A.1, 2007.
Davies, G. R., Cliff, R. A., Norry, M. J., and Gerlach, D. C.: A combined chemical and Pb-Sr-Nd isotope study of the Azores and Cape Verde hot-spots: The geodynamic implications, Geol. Soc. Spec. Publ., 42, 231–255, https://doi.org/10.1144/GSL.SP.1989.042.01.15, 1989.
Dingwell, D. B., O'Neill, H. S. C., Ertel, W., and Spettel, B.: The solubility and oxidation state of nickel in silicate melt at low oxygen fugacities: Results using a mechanically assisted equilibration technique, Geochim. Cosmochim. Ac., 58, 1967–1974, https://doi.org/10.1016/0016-7037(94)90428-6, 1994.
Feig, S. T., Koepke, J., and Snow, J. E.: Effect of water on tholeiitic basalt phase equilibria: An experimental study under oxidizing conditions, Contrib. Mineral. Petr., 152, 611–638, https://doi.org/10.1007/s00410-006-0123-2, 2006.
Feig, S. T., Koepke, J., and Snow, J. E.: Effect of oxygen fugacity and water on phase equilibria of a hydrous tholeiitic basalt, Contrib. Mineral. Petr., 160, 551–568, https://doi.org/10.1007/s00410-010-0493-3, 2010.
Ferraris, G. and Ivaldi, G.: Structural features of micas, Rev. Mineral. Geochem., 46, 117–153, https://doi.org/10.2138/rmg.2002.46.03, 2002.
Fiorentini, M. L., Beresford, S. W., Stone, W. E., and Deloule, E.: Evidence of water degassing during emplacement and crystallization of 2.7 Ga komatiites from the Agnew-Wiluna greenstone belt, Western Australia, Contrib. Mineral. Petr., 164, 143–155, https://doi.org/10.1007/s00410-012-0730-z, 2012.
Foley, S. F.: A reappraisal of redox melting in the earth's mantle as a function of tectonic setting and time, J. Petrol., 52, 1363–1391, https://doi.org/10.1093/petrology/egq061, 2011.
Fonseca, R. O., Mallmann, G., Sprung, P., Sommer, J. E., Heuser, A., Speelmanns, I. M., and Blanchard, H.: Redox controls on tungsten and uranium crystal/silicate melt partitioning and implications for the U/W and Th/W ratio of the lunar mantle, Earth Planet. Sc. Lett., 404, 1–13, 2014.
Frost, D. J. and McCammon, C. A.: The Redox State of Earth's Mantle, Annu. Rev. Earth Pl. Sc., 36, 389–420, https://doi.org/10.1146/annurev.earth.36.031207.124322, 2008.
Giesting, P. A. and Filiberto, J.: The formation environment of potassic-chloro-hastingsite in the nakhlites MIL 03346 and pairs and NWA 5790: Insights from terrestrial chloro-amphibole, Meteorit. Planet. Sci., 51, 2127–2153, https://doi.org/10.1111/maps.12675, 2016.
Green, D. H., Hibberson, W. O., Rosenthal, A., Kovács, I., Yaxley, G. M., Falloon, T., and Brink, F.: Experimental study of the influence of water on melting and phase assemblages in the upper mantle, J. Petrol., 55, 2067–2096, https://doi.org/10.1093/petrology/egu050, 2014.
Griffin, W. L., Powell, W., Pearson, N. J., and O'reilly, S. Y.: GLITTER: data reduction software for laser ablation ICP-MS, Laser Ablation ICP-MS in the Earth Sciences: Current practices and outstanding issues, 308–311, 2008.
Hawthorne, F. C. and Oberti, R.: Amphiboles: Crystal Chemistry, Rev. Mineral. Geochem., 67, 1–54, https://doi.org/10.2138/rmg.2007.67.1, 2007.
Hofmann, A. W.: Mantle geochemistry: the message from oceanic volcanism, Nature, 385, 219–229, https://doi.org/10.1038/385219a0, 1997.
Jenkins, D. M.: The incorporation of chlorine into calcium amphibole, Am. Mineral., 104, 514–524, https://doi.org/10.2138/am-2019-6768, 2019.
Jenner, F. E., O'Neill, H. S. C., Arculus, R. J., and Mavrogenes, J. A.: The magnetite crisis in the evolution of arc-related magmas and the initial concentration of Au, Ag and Cu, J. Petrol., 51, 2445–2464, https://doi.org/10.1093/petrology/egq063, 2010.
Johannes, W. and Bode, B.: Loss of iron to the Pt-container in melting experiments with basalts and a method to reduce it, Contrib. Mineral. Petr., 67, 221–225, https://doi.org/10.1007/BF01046578, 1978.
King, P. L., Hervig, R. L., Holloway, J. R., Vennemann, T. W., and Righter, K.: Oxy-substitution and dehydrogenation in mantle-derived amphibole megacrysts, Geochim. Cosmochim. Ac., 63, 3635–3651, https://doi.org/10.1016/S0016-7037(99)00162-3, 1999.
Lange, R. A. and Carmichael, I. S. E.: A potassic volcanic front in western Mexico: the lamprophyric and related lavas of San Sebastian, Geol. Soc. Am. Bull., 103, 928–940, 1991.
Lee, C. T. A., Leeman, W. P., Canil, D., and Li, Z. X. A.: Similar V/Sc systematics in MORB and arc basalts: Implications for the oxygen fugacities of their mantle source regions, J. Petrol., 46, 2313–2336, https://doi.org/10.1093/petrology/egi056, 2005.
Li, X., Zhang, C., Behrens, H., and Holtz, F.: Calculating amphibole formula from electron microprobe analysis data using a machine learning method based on principal components regression, Lithos, 362–363, 105469, https://doi.org/10.1016/j.lithos.2020.105469, 2020.
Luth, R. W: Natural versus experimental control of oxidation-state-effects on the composition and speciation of C-O-H fluids, Am. Mineral., 74, 50–57, 1989.
Makino, K., Tomita, K., and Suwa, K.: Effect of chlorine on the crystal structure of a chlorine-rich hastingsite, Mineral. Mag., 57, 677–685, https://doi.org/10.1180/minmag.1993.057.389.12, 1993.
Matjuschkin, V., Brooker, R. A., Tattitch, B., Blundy, J. D., and Stamper, C. C.: Control and monitoring of oxygen fugacity in piston cylinder experiments, Contrib. Mineral. Petr., 169, 1–16, https://doi.org/10.1007/s00410-015-1105-z, 2015.
McCammon, C.: The Paradox of Mantle Redox, Science, 308, 807–808, https://doi.org/10.1126/science.1110532, 2005.
Menzies, M. and Murthy, R. V.: Nd and Sr isotope geochemistry of hydrous mantle nodules and their host alkali basalts: implications for local heterogeneities in metasomatically veined mantle, Earth Planet. Sc. Lett., 46, 323–334, 1980.
Müntener, O. and Ulmer, P.: Experimentally derived high-pressure cumulates from hydrous arc magmas and consequences for the seismic velocity structure of lower arc crust, Geophys. Res. Lett., 33, 1–5, https://doi.org/10.1029/2006GL027629, 2006.
Nandedkar, R.H., Hürlimann, N., Ulmer, P., and Müntener, O.: Amphibole – melt trace element partitioning of fractionating calc-alkaline magmas in the lower crust: an experimental study, Contrib. Mineral. Petr., 171, 1–25, https://doi.org/10.1007/s00410-016-1278-0, 2016.
Oberti, R., Ungaretti, L., Cannillo, E., and Hawthorne, F. C.: The mechanism of Cl incorporation into amphibole, Am. Mineral., 78, 746–752, 1993.
Oberti, R., Hawthorne, F. C., Cannillo, E., and Cámara, F.: Long-Range Order in Amphiboles, Rev. Mineral. Geochem., 67, 125–171, https://doi.org/10.2138/rmg.2007.67.4, 2007.
Pilet, S., Baker, M. B., and Stolper, E. M.: Metasomatized lithosphere and the origin of alkaline lavas, Science, 320, 916–919, https://doi.org/10.1126/science.1156563, 2008.
Pirard, C. and Hermann, J.: Experimentally determined stability of alkali amphibole in metasomatised dunite at sub-arc pressures, Contrib. Mineral. Petr., 169, 1, https://doi.org/10.1007/s00410-014-1095-2, 2015.
Popp, R. K., Virgo, D., Yoder, H. S., Hoering, T. C., and Phillips, M. W.: An experimental study of phase equilibria and Fe oxy-component in kaersutitic amphibole: implications for the fH2 and aH2O in the upper mantle, Am. Mineral., 80, 534–548, https://doi.org/10.2138/am-1995-5-613, 1995.
Popp, R. K., Hibbert, H. A., and Lamb, W. M.: Oxy-amphibole equilibria in Ti-bearing calcic amphiboles: Experimental investigation and petrologic implications for mantle-derived amphiboles, Am. Mineral., 91, 54–66, https://doi.org/10.2138/am.2006.1838, 2006.
Rustioni, G., Audétat, A., and Keppler, H.: Experimental evidence for fluid-induced melting in subduction zones, Geochem. Perspect. Lett., 11, 49–54, 2019.
Scambelluri, M., Cannaò, E., and Gilio, M.: The water and fluid-mobile element cycles during serpentinite subduction. A review, Eur. J. Mineral., 31, 405–428, https://doi.org/10.1127/ejm/2019/0031-2842, 2019.
Shannon, R. D.: Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr. Sect., 32, 751–767, 1976.
Sisson, T. W., Ratajeski, K., Hankins, W. B., and Glazner, A. F.: Voluminous granitic magmas from common basaltic sources, Contrib. Mineral. Petr., 148, 635–661, https://doi.org/10.1007/s00410-004-0632-9, 2005.
Smith, D. J.: Clinopyroxene precursors to amphibole sponge in arc crust, Nat. Commun., 5, 1–6, https://doi.org/10.1038/ncomms5329, 2014.
Stamper, C. C., Melekhova, E., Blundy, J. D., Arculus, R. J., Humphreys, M. C. S., and Brooker, R. A.: Oxidised phase relations of a primitive basalt from Grenada, Lesser Antilles, Contrib. Mineral. Petr., 167, 1–20, https://doi.org/10.1007/s00410-013-0954-6, 2014.
Tiepolo, M., Vannucci, R., Bottazzi, P., Oberti, R., Zanetti, A., and Foley, S.: Partitioning of rare earth elements, Y, Th, U, and Pb between pargasite, kaersutite, and basanite to trachyte melts: Implications for percolated and veined mantle, Geochem., Geophy. Geosy., 1, 1039m https://doi.org/10.1029/2000GC000064, 2000a.
Tiepolo, M., Vannucci, R., Oberti, R., Foley, S., Bottazzi, P., and Zanetti, A.: Nb and Ta incorporation and fractionation in titanian pargasite and kaersutite: Crystal-chemical constraints and implications for natural systems, Earth Planet. Sc. Lett. 176, 185–201, https://doi.org/10.1016/S0012-821X(00)00004-2, 2000b.
Tiepolo, M., Oberti, R., Zanetti, A., Vannucci, R., and Foley, S. F.: Trace-Element Partitioning Between Amphibole and Silicate Melt, Rev. Mineral. Geochem., 67, 417–452, https://doi.org/10.2138/rmg.2007.67.11, 2007.
Tiepolo, M., Tribuzio, R., Langone, A., Di, D., Della, S., and Dell, T. E.: High-Mg Andesite Petrogenesis by Amphibole Crystallization and Ultramafic Crust Assimilation: Evidence from Adamello Hornblendites (Central Alps, Italy), J. Petrol., 52, 1011–1045, https://doi.org/10.1093/petrology/egr016, 2011.
Ulmer, P. and Luth, R. W.: The graphite-COH fluid equilibrium in P, T, fO2 space – An experimental determination to 30 kbar and 1600 ∘C, Contrib. Mineral. Petr., 106, 265–272, https://doi.org/10.1007/BF00324556, 1991.
Ulmer, P., Kaegi, R., and Müntener, O.: Experimentally derived intermediate to silica-rich arc magmas by fractional and equilibrium crystallization at 1⋅0 GPa: An evaluation of phase relationships, compositions, liquid lines of descent and oxygen fugacity, J. Petrol., 59, 11–58, https://doi.org/10.1093/petrology/egy017, 2018.
Van den Bleeken, G. and Koga, K. T.: Experimentally determined distribution of fluorine and chlorine upon hydrous slab melting, and implications for F–Cl cycling through subduction zones, Geochim. Cosmochim. Ac., 171, 353–373, 2015.
Wood, B. J. and Blundy, J. D.: 3.11 Trace Element Partitioning: The Influences of Ionic Radius, Cation Charge, Pressure, and Temperature, Treatise on Geochemistry, 2nd Edn., Elsevier, https://doi.org/10.1016/B978-0-08-095975-7.00206-0, 2014.
- Abstract
- Introduction
- Experimental and analytical techniques
- Experimental results
- Attainment of equilibrium
- Trace element partitioning between amphibole and glass (D)
- Discussion
- General implications and conclusions
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Experimental and analytical techniques
- Experimental results
- Attainment of equilibrium
- Trace element partitioning between amphibole and glass (D)
- Discussion
- General implications and conclusions
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement