the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Apr 2026
| 20 Apr 2026
Selective uptake of rare earth elements and other cations in sector-zoned natural calcite as analogues for trivalent actinide behavior
Ferdinand Baumeister
Martin Kutzschbach
Martina Klinkenberg
Felix Brandt
Thomas Neumann
Calcite has been shown to incorporate trivalent actinides into its crystal lattice, highlighting its potential to contribute to radionuclide retention processes in the environment. Earlier studies on calcite's elemental uptake were conducted under controlled laboratory conditions, which do not fully capture the complexities of natural environments. To gain a deeper understanding of calcite's uptake capacity under natural conditions, a sample from the Wenzel ore mine, Germany, was analyzed, originating from a calcite vein that formed under conditions relevant to deep geological-waste repositories. The chemical analogy between rare earth elements (REEs) and trivalent actinides helps to evaluate the retention potential and incorporation mechanisms of trivalent radionuclides. A micro-X-ray fluorescence (µXRF) element map revealed that the investigated calcite consists of an euhedral crystal core exhibiting sector zoning, characterized by trace element heterogeneity across coevally grown crystal faces. High-resolution laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) further refined and quantified the elemental distribution, revealing preferential incorporation of specific elements, particularly REEs, within one sector of the calcite. Remarkably, this sector showed REE concentrations over 200 times higher than those in the depleted sector, indicating a significant potential for the retention of trivalent actinides. Furthermore, the data indicate that charge equilibration of incorporated trivalent cations occurs via two processes: coupled substitution with monovalent cations and vacancies in the crystal structure. Overall, these results demonstrate that sector-zoned calcite formed under repository-relevant conditions can maintain high retention potential for trivalent actinides, even in environments depleted in monovalent cations.
- Article
(6011 KB) - Full-text XML
-
Supplement
(464 KB) - BibTeX
- EndNote




The environmental challenges associated with the operation of nuclear power plants and the resulting generation of high-level radioactive waste necessitate effective long-term disposal solutions. Currently, the safest approach is considered to be disposal in deep geological-waste repositories (e.g., Kim et al., 2011; Birkholzer et al., 2012; Kurniawan et al., 2021). When selecting suitable repository sites, it is essential to minimize risks that could lead to radionuclide release from the waste canisters into the environment. Modern repository designs consist of a multi-barrier system (Olszewska et al., 2015; Apted and Ahn, 2017) to ensure the long-term safety of the repository. Nevertheless, within the required time frames of up to 1×106 years evaluated in safety assessments, the possibility of radionuclide migration and release into the environment cannot be entirely excluded. It is therefore essential to assess potential host rocks with respect to radionuclide retention.
In the event of canister corrosion and subsequent release of trivalent actinides (An3+) from spent nuclear fuel into the near field of the repository, interactions with carbonate crystals within the host rock formations can function as a retention mechanism by structural uptake and solid-solution formation. Curti (1999) identified calcite as an effective scavenger for An3+ and reported its high partitioning behavior based on empirical correlations. However, only natural samples can accurately replicate the conditions found in natural systems, which include the variability of physicochemical parameters across geological timescales. Given the limited availability of radionuclides in natural samples, inactive natural analogues become valuable tools for the investigation of the potential behavior of radionuclides within such systems. The analogy between An3+ and rare earth elements (REEs) has been described by various studies (e.g., Krauskopf, 1986; Bruno and Sandino, 1987; Zhong and Mucci, 1995; Jensen and Bond, 2002; Nedel et al., 2010; Heller et al., 2012; Heller et al., 2023) and led to various studies on the incorporation behavior of REE into calcite (e.g., Lakshtanov and Stipp, 2004; Stipp et al., 2009; Gabitov et al., 2017; Drake et al., 2017).
The structural complexity of calcite, characterized by distinct faces with varying surface energies and coordination environments, significantly influences trace element incorporation. Several crystallographic faces are commonly exhibited by calcite, including the thermodynamically stable (104) face, which is the most abundant and dominant cleavage plane, along with less common faces such as (110), (012), and (001) (Heberling et al., 2011; 2014). The rhombohedral structure of calcite exhibits obtuse and acute faces, which differ in terms of their surface atom arrangements and bonding characteristics (Paquette and Reeder, 1995). During calcite growth via spiral mechanisms on these faces (Reeder, 1996), distinct step edges with different atomic arrangements create preferential incorporation sites for various cations. To enhance the predictive capacity regarding the retention of An3+ in natural calcite, a comprehensive understanding of the structural uptake mechanisms is essential. The phenomenon of face-dependent partitioning in calcite, commonly referred to as sector zoning, was first identified by Reeder and Paquette (1989), who observed the varying accumulation of elements such as Mn, Sr, and Fe within distinct concurrently grown sectors of calcite. They assumed that the partitioning of these elements into specific sectors is influenced by factors including ionic size, charge, and electron configuration. A subsequent study by Reeder (1996) expanded this investigation to include additional elements, namely Mn, Sr, Ba, Cd, Co, and Zn, providing factors of preferred incorporation into an enriched sector compared to a depleted one that ranged from 2 to 10. Furthermore, Barker and Cox (2011) demonstrated that REEs are also subject to sector zoning in calcite. Kusturica et al. (2022) explored the trace element (TE) uptake in natural calcite, revealing a significant retention potential for REEs, and confirmed the occurrence of face-dependent partitioning through the application of laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS).
The incorporation of trivalent cations into the calcite lattice requires charge equilibration, which can be achieved through two primary mechanisms: coupled substitution involving a monovalent cation or the presence of a vacancy adjacent to two incorporated trivalent cations. The coupled substitution mechanism, where REE3+ incorporation into calcite is charge-balanced by co-substitution with monovalent cations (e.g., Na+), is strongly supported by experimental and spectroscopic evidence. Studies such as those by Marques Fernandes et al. (2008), Voigt et al. (2017), and Möller and De Lucia (2020) demonstrate that Na+ plays a critical role in stabilizing REE3+ within the calcite lattice via the following exchange reaction: 2Ca2+ ⇌ REE3+ + Na+. This mechanism dominates in environments with elevated monovalent cation concentrations, such as marine or hydrothermal systems, where Na+ availability facilitates charge compensation. However, Gabitov et al. (2017) observed reduced REE partitioning in low-Na+ solutions, highlighting the dependency of this pathway on solution chemistry. The incorporation efficiency varies with REE ionic radii, favoring lighter REE due to their closer match to Ca2+.
The vacancy model, where two REE3+ ions substitute for three Ca2+ ions while creating a lattice vacancy, provides an alternative pathway under monovalent cation-deficient conditions. Lakshtanov and Stipp (2004) showed that Eu incorporation aligns with this mechanism, particularly at low Na+ concentrations, as the vacancy compensates for charge imbalance without requiring additional cations. Perry (2019) emphasized that this model becomes thermodynamically favorable when structural strain from REE3+ substitution is minimized, though it is less efficient than coupled substitution due to higher lattice distortion. Experimental data confirm its relevance in low-salinity or high-pH environments, where REE–hydroxyl complexes may dominate aqueous speciation. While vacancy-mediated uptake preserves REE signatures in calcite, it exhibits stronger fractionation between light and heavy REEs compared to coupled substitution.
This study investigates REE incorporation as analogues for trivalent actinides in natural calcite, with specific emphasis on (1) spatial distribution patterns and sector-specific partitioning behavior, (2) the quantitative assessment of retention efficiency across different crystal faces, and (3) the evaluation of dominant charge equilibration mechanisms under repository-relevant conditions. For this purpose, a calcite crystal originating from the tunnel wall of the Wenzel mine (Schwarzwald Mountains, Germany) was selected. The crystal precipitated during a low-temperature hydrothermal episode (50–70 °C) (Burisch et al., 2018). According to the distance from a repository and the stage of radioactive decay of the waste, a relevant share of the repository surrounding the host rock is characterized by these temperature conditions (Zhao et al., 2014; Xue et al., 2021). µXRF is employed in combination with scanning electron microscopy (SEM) and LA-ICP-MS to achieve high-spatial-resolution element maps of trace element distributions in the sampled calcite, with a particular focus on the heterogeneous uptake across different calcite sectors. The findings from this study will contribute to a better understanding of the trace element incorporation patterns, as well as an improved evaluation of the retention and remobilization potential of radionuclides in natural calcite.
The Schwarzwald Mountains in the southwest of Germany have been the object of various research campaigns and are therefore geologically well explored (Schwinn and Markl, 2005; Hann, 2006; Geyer et al., 2011; Staude et al., 2012). Located in the Moldanubian Zone of the Central European Variscan fold belt (Kalt et al., 1995, 2000), the Schwarzwald Mountains are mainly composed of gneisses and Varsican granites, as well as minor occurrences of retrograded peridotites, pyroxenites, eclogites, and amphibolites (Kalt et al., 2000). The predominantly S-type granites have been dated to an intrusion age between 335 and 315 Ma (Kalt et al., 2000), unconformably overlain by Permian arkoses and Triassic sandstones and conglomerates, specifically the Bunter Sandstone. During the Rhine Graben uplift, a continental rift structure (Pflug, 1982) led to erosion of these rocks, exposing the basement rocks to the surface.
The Schwarzwald mining district has been exploited since Neolithic and Roman times (Werner and Dennert, 2004) and hosts over 400 hydrothermal vein-type deposits. Classification based on fluid inclusion characteristics, geochemical disparities, and mineralization style (Baatartsogt et al., 2007) categorizes the Schwarzwald veins into quartz veins with Sb ± Au ± Ag mineralization, likely Variscan in age, and post-Variscan fluorite–barite–quartz or carbonate veins containing Fe, Mn, Ag, Co, Ni, U, Cu, Pb, or Zn mineralization (Metz et al., 1957). Euhedral carbonates represent the latest hydrothermal phases in vugs and are characterized as forming late and under relatively low temperatures compared to the older mineralization within the veins (Burisch et al., 2018). This late-stage calcite formation is presumed to have a hydrothermal origin, supported by fluid inclusion studies indicating low salinity and homogenization temperatures between 50 and 60 °C (Walter et al., 2015). Burisch et al. (2018) conducted a study on the youngest mineralization event in the Schwarzwald mining district, identifying three subsequent generations of the late-stage vein assemblage with U–Pb carbonate ages between 20 and 0.6 Ma. Within the Wenzel mine, the only age dating data are based on a dolomite mineral at 8 Ma and a younger calcite at 0.6 Ma. In contrast, calcite samples from nearby locations (Artenberg quarry; 10 km from Wenzel mine) yielded ages of 20 and 14 Ma (Burisch et al., 2018). The authors also conducted a thermodynamic modeling approach, suggesting uplift-related fluid mixing of deeply sourced Mg-, Fe-, and SO4-rich fluid with shallow crustal HCO3-rich fluids. Increasing shares of shallow waters with a temperature between 50 and 70 °C are assumed to take place in the fluid-mixing situation in surface close formations. The prevailing conditions reflect a similar temperature regime within the near-field of a nuclear waste repository, making them an ideal setting to investigate trace element uptake into fracture-filling minerals.
The Wenzel mine, situated near Oberwolfach in the central Schwarzwald Mountains, is a historic silver ore mine first documented in the year 1700. Over time, the mine naturally flooded with groundwater in its deeper tunnels until its reopening around the year 2000, being accompanied by drainage of the tunnel system. A calcite vein of hydrothermal origin located in the “tiefe Strecke” section of the mine hosted crystals, predominantly exhibiting euhedral shapes, located directly at the tunnel walls. One crystal, which was exposed with one face to the tunnel wall, was sampled for further investigation and exhibited a size of approx. 1.5 cm × 2 cm. The morphological similarities – such as colorlessness, transparency, size, and scalenohedral crystal morphology – between the 20 Myr old samples from the nearby Artenberg quarry (Burisch et al., 2018) and the sample of the present study suggest that the calcite core of this sample corresponds to hydrothermal calcite precipitation at these ages. The crystal was mounted in a 1 in. epoxy resin mount and polished for subsequent analyses.
4.1 Scanning electron microscopy
The environmental scanning electron microscope Quanta 200 F (FEI, Eindhoven, the Netherlands) was used for detailed microstructural analysis of the sample. Backscattered-electron (BSE) images, as well as secondary electron (SE) images, were taken at a high voltage of 20 kV, a spot size of 4, and a working distance of 10 mm in low-vacuum mode at 60 Pa.
4.2 Micro X-ray fluorescence
µXRF mapping was carried out on the polished calcite mount using a Bruker M4 Tornado at the MAGMA Lab of the Technical University Berlin. The instrument is equipped with a capillary X-ray optic and two 30 mm2 silicon drift detectors. Measurements were performed with a 20 µm beam diameter; the Rh anode was run at 50 kV and 600 µA under a chamber vacuum of 20 mbar. Element maps were acquired on a 20 µm grid with a dwell time of 40 ms per pixel, yielding a total acquisition time of roughly 18 h for the entire thin section. The X-ray tube output and the positions of the characteristic fluorescence lines are checked and adjusted on a monthly basis using Bruker single-element reference materials. These measurements provided semi-quantitative distributions of major and trace elements and were used to select representative areas for subsequent quantitative analysis by LA-ICP-MS.
4.3 Laser ablation ICP-MS mapping
High-resolution elemental maps were acquired in the MAGMA Lab (TU Berlin) with an Agilent 8900 ICP-MS/MS coupled to a 193 nm Analyte Excite excimer laser (Teledyne Photon Machines). Helium served as a carrier gas (total 0.95 L min−1; 0.50 L min−1 through the cell and 0.45 L min−1 through the cup).
After plasma ignition and initialization of the He flow, signals at = 42 (e.g., 14N14N14N+) and = 31 (e.g., 15N16O+) were monitored to track residual air in the cell and transfer lines. Once these background signals had stabilized at < 20 000 cps, the Ar nebulizer and He carrier flows were adjusted while ablating NIST SRM 610 in line scan mode, targeting () < 0.05 % and a ratio () of 100 ± 1 %. To further increase sensitivity, 3–4 mL min−1 of 99.999 % N2 was added to the Ar nebulizer gas. Under these conditions, an automatic lens optimization was performed on NIST 610 to maximize signal intensity in the mid-mass range.
The aerosol rapid introduction system (ARIS; Teledyne Photon Machines) was used for fast signal transport. When mapping more than four elements simultaneously, a low-volume glass expansion adapter was installed in the interface to increase the single-pulse response (combined wash-in–wash-out). The single-pulse response (SPR) was characterized by ablating NIST 610 at 1 Hz and dosage 1 in line scan mode, monitoring (U) with a dwell time of 5 ms.
By averaging the peak shape of 300 single pulses, the SPR was determined at the full width at 10 % of the maximum intensity. The SPR was found to be between 30 ms (without adapter) and 120 ms (with adapter).
For the actual maps, the following mass channels were recorded: (Mg), (Ca), (Mn), (Fe), (Sr), (La), (Ce), and (Yb).
The crystal segment was rastered using unidirectional scan lines without line overlap, with a 2 s delay between lines to minimize memory effects. The laser fluence was 3.5 J cm−2, the spot size was 3 µm, the applied dosage was 10 (corresponding to a 10-fold spot overlap), and the repetition rate was 133 Hz. These settings were chosen to avoid aliasing artifacts. Dwell times for each isotope were allocated based on preliminary test lines to ensure signal-to-noise ratios above 20. The total sweep time was tuned to prevent aerosol carry-over beyond the laser spot diameter during scanning. The resulting ablation depth was on the order of approx. 1–2 µm for the selected dosage.
Raw data reduction and image reconstruction were carried out in HDIP (Teledyne Photon Machines). Background correction was performed using a cubic spline function. Quantitative maps were obtained by calibrating each pixel against repeated (≥ 10) line scans of NIST SRM 612, recorded immediately before and after mapping the target grain. Subsequently, two-dimensional regions within calcite were defined, and a normalization factor was derived such that the mean Ca concentration in these regions matched that of stoichiometric CaCO3 (Ca = 40.04 wt %). This normalization factor was then uniformly applied to all pixels of the map. Instrumental drift was tracked and corrected using the NIST 612 line scans but was usually negligible.
4.4 Laser ablation ICP-MS spot analysis
Because the very small laser spot sizes and short dwell times used for elemental mapping constrain both detection limits and the number of isotopes that can be acquired per sweep, additional LA-ICP-MS spot measurements with a larger spot diameter of 50 µm were carried out on the same sample after completion of the maps. For these spot analyses, the repetition rate was reduced to 10 Hz, and a large-volume adapter from glass expansion was installed to increase signal stability. The following mass channels were monitored: (Li), (Be), (Na), (Mg), (P), (K), (Ca), (Sc), (V), (Cr), (Mn), (Fe), (Co), (Ni), (Cu), (Zn), (As), (Se), (Rb), (Sr), (Y), (Zr), (Nb), (Mo), (Cd), (Sb), (Ba), (La), (Ce), (Pr), (Nd), (Sm), (Eu), (Gd), (Tb), (Dy), (Ho), (Er), (Tm), (Yb), (Lu), (Hf), (Ta), (W), (Pb), (Th), and (U). Each spot analysis comprised 30 s gas blank acquisition, 40 s laser ablation, and a 30 s wash-out period. The total sweep time per integration cycle was 0.4928 s, and the laser fluence at the sample surface was set to 4.25 J cm−2 . All elements were quantified against NIST SRM 612 using Ca as an internal standard, and the resulting time-resolved signals were processed with the HDIP software. The results in µg g−1 and mol g−1, including 2SD uncertainties, are presented in the Supplement (data 1). Additionally, the calcite powder standard ECRM752-NP was used to quantify matrix effects. Deviations from the certified concentrations were calculated for the elements provided by the standard material, showing good general agreement. Most elements match within analytical uncertainties, with MREE and HREE exhibiting slight deviations of < 22 % (Supplement – data 2). Notable outliers are Y (51 %) and Eu (93 %), and their results should therefore be interpreted with caution. ECRM752-NP provides the closest available matrix match, as no homogeneous natural calcite reference material currently exists to our knowledge. Although absolute quantification of matrix effects is not possible, the results of this study can be regarded to be accurate to within the stated uncertainties, with minor deviations for MREE and HREE.
5.1 Calcite growth structure
To identify growth structures of the calcite crystal, a µXRF multi-element mapping was conducted (Fig. 1, qualitative measurement). The spatial distribution of Mn reveals two primary growth structures within the mineral: a core characterized by relatively low Mn concentration (Fig. 1, labeled as “core”) and Mn-enriched overgrowth zones. The crystal core (approx. 6 mm × 12 mm) exhibits an almost euhedral morphology and shows distinct sector zoning, reflecting compositional heterogeneities in Mn distribution. The larger sector, which displays a higher Mn content, is designated as sector A, while the smaller sector, characterized by lower Mn content, is referred to as sector B (Fig. 1). The overgrowth zones have elevated Mn concentrations compared to the core, resulting in a layered structure that aligns with the euhedral crystal faces of the core. These outer zones exhibit variable thicknesses ranging from 0.1 to 1 mm. The subsequent zone features undulate structures and shows signs of dissolution and reprecipitation. This outermost overgrowth zone attains a maximum width of 2 mm and is partially lashed out in some regions of the crystal. The present paper focuses on the sector zonation of the euhedral crystal core as it is assumed to represent temperature conditions comparable to the regime near the repository. The overgrown calcite layers are suspected to be formed under lower temperature conditions in the groundwater.
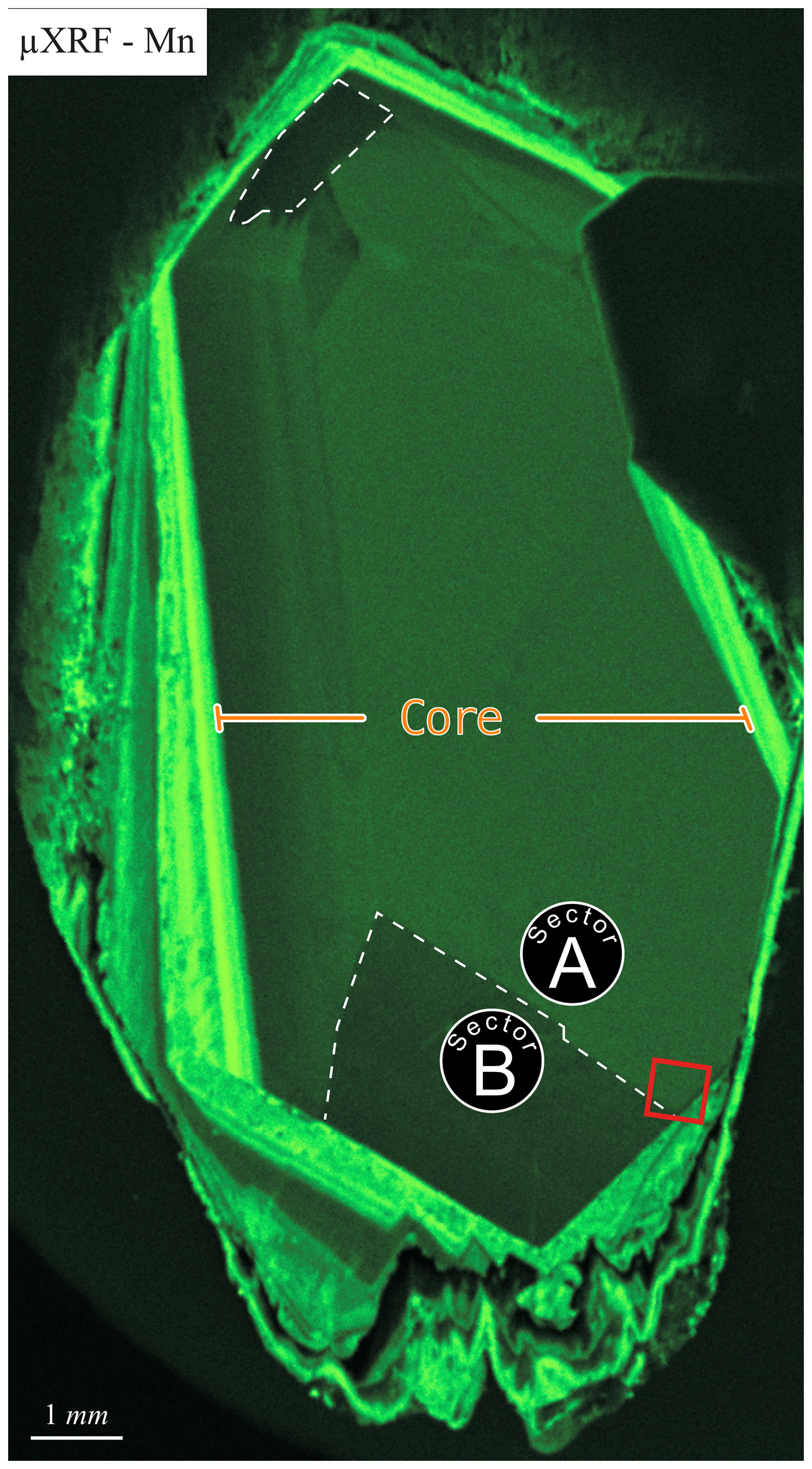
Figure 1Mn distribution in the sample identified by µXRF. The core segment of the calcite is indicated by the orange marker. Dashed white lines mark sectors A and B within the core. The red rectangle indicates the location of the subsequent LA-ICP-MS element map.
5.2 Chemical unconformity of sector zones within the core
A comprehensive understanding of sector zonation growth necessitates a higher spatial resolution and detailed concentration data for a broader range of elements. To address this requirement, an area was selected that includes both sectors A and B (Fig. 1, red area). The SEM imaging revealed a distinct contrast between sectors A and B (Fig. 2a), with sector B appearing to be brighter than sector A. The SEM contrast correlates well with the elemental distribution of the Mn identified via µXRF. The LA-ICP-MS element distribution maps reveal a distinct tendency of each analyzed trace element to concentrate in one of the two sectors (Fig. 2b–h). Within both sectors, a predominantly homogeneous distribution of elements is evident. An exception is a slight Mn depletion located in a halo structure (Fig. 2b, red area), which is spatially associated with two linear features. One linear feature is visible in the SEM image (Fig. 2a; green arrow), while a second, subparallel linear structure is only expressed in the La and Ce distribution maps as a sharp change in concentration along a straight, cross-cutting line (Fig. 2g, h; green arrow). The distribution of Mn, as illustrated in Fig. 2b, aligns with previous µXRF element map results and underscores the preferential incorporation of Mn into sector A, with averaged concentration values of 5550 ± 120 µg g−1, compared to 4160 ± 60 µg g−1 in sector B. This trend is similarly observed for Mg and Fe, where sector A exhibits higher concentrations (Mg: 68.2 ± 3.0 µg g−1, Fe: 176.6 ± 5.1 µg g−1) relative to sector B (Mg: 44.2 ± 1.1 µg g−1, Fe: 133.1 ± 5.8 µg g−1). Conversely, elements such as Sr, Yb, La, and Ce demonstrate a preference for sector B, with higher concentrations observed (Sr: 75.0 ± 0.9 µg g−1, Yb: 32.6 ± 0.7 µg g−1, La: 8.49 ± 0.27 µg g−1, Ce: 28.6 ± 0.3 µg g−1) compared to sector A (Sr: 61.5 ± 1.5 µg g−1, Yb: 0.36 ± 0.13 µg g−1, La: 0.038 ± 0.036 µg g−1, Ce: 0.13 ± 0.10 µg g−1). Table 1 summarizes the concentration factors between the two sectors, highlighting the preferential distribution of these elements. Remarkably, the La and Ce concentration levels in sector B exceed the values in sector A by a factor of over 200 (Table 1). LA-ICP-MS spot analyses were conducted as indicated in Fig. 3a to obtain additional information for a broader range of elements, with spots 1, 2, 3, and 4 corresponding to sector A and spots 5, 6, and 7 corresponding to sector B (data are provided in the Supplement – data 1). The REE concentration distribution derived from this method clearly indicates a preference for higher uptake in sector B (Fig. 4). Both sectors exhibit a similar REE distribution pattern characterized by constant light-REE (LREE) and intermediate-REE (MREE) concentrations and slightly increasing heavy-REE (HREE) concentrations normalized to chondrite. Furthermore, a negative Eu anomaly is present, as calculated using the formula with Eu* .
Table 1Factor of element concentration between sectors A and B. For calculation, spot analyses 1, 2, 3, and 4 (sector A) and 5, 6, and 7 (sector B) were considered.
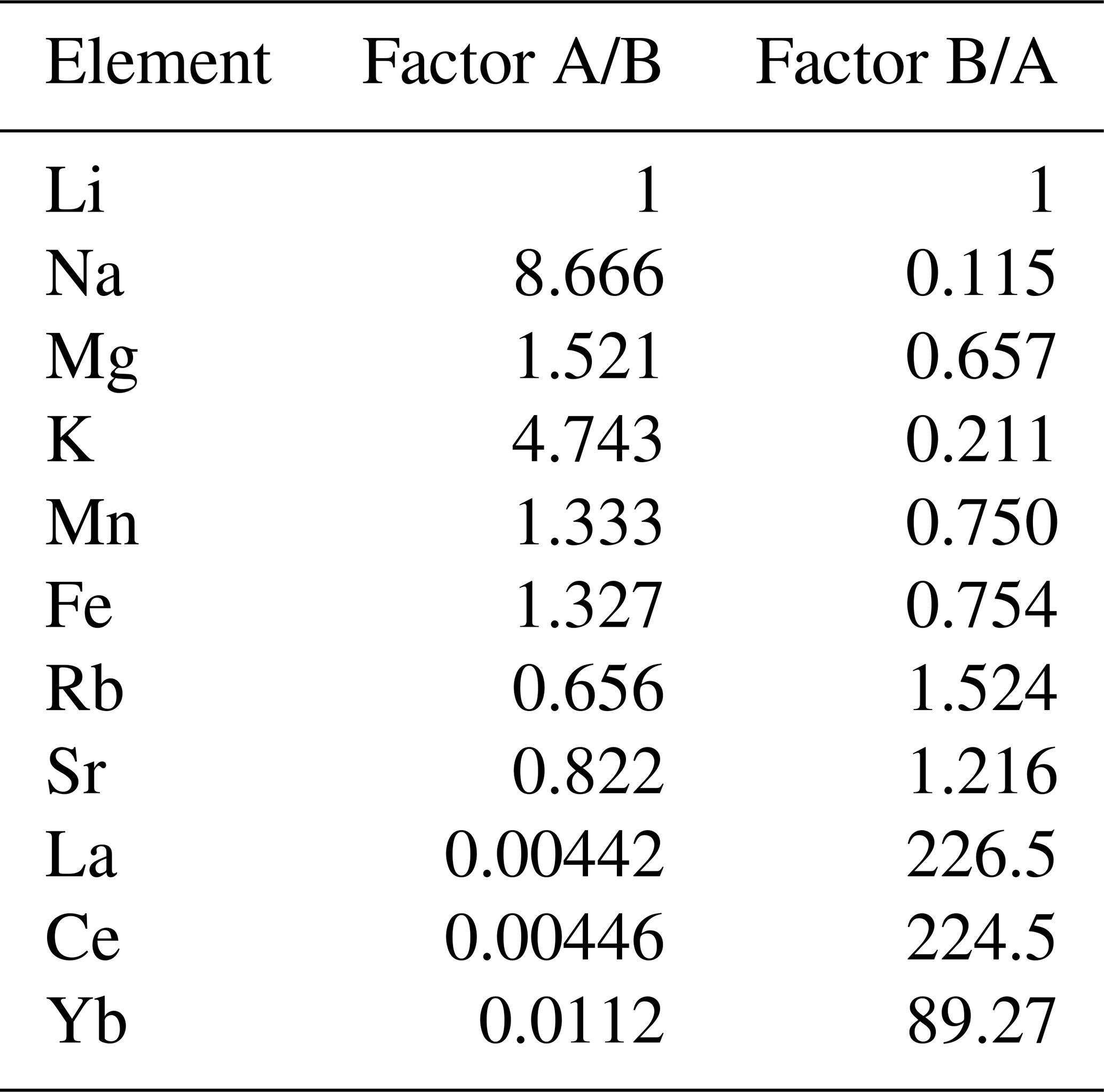
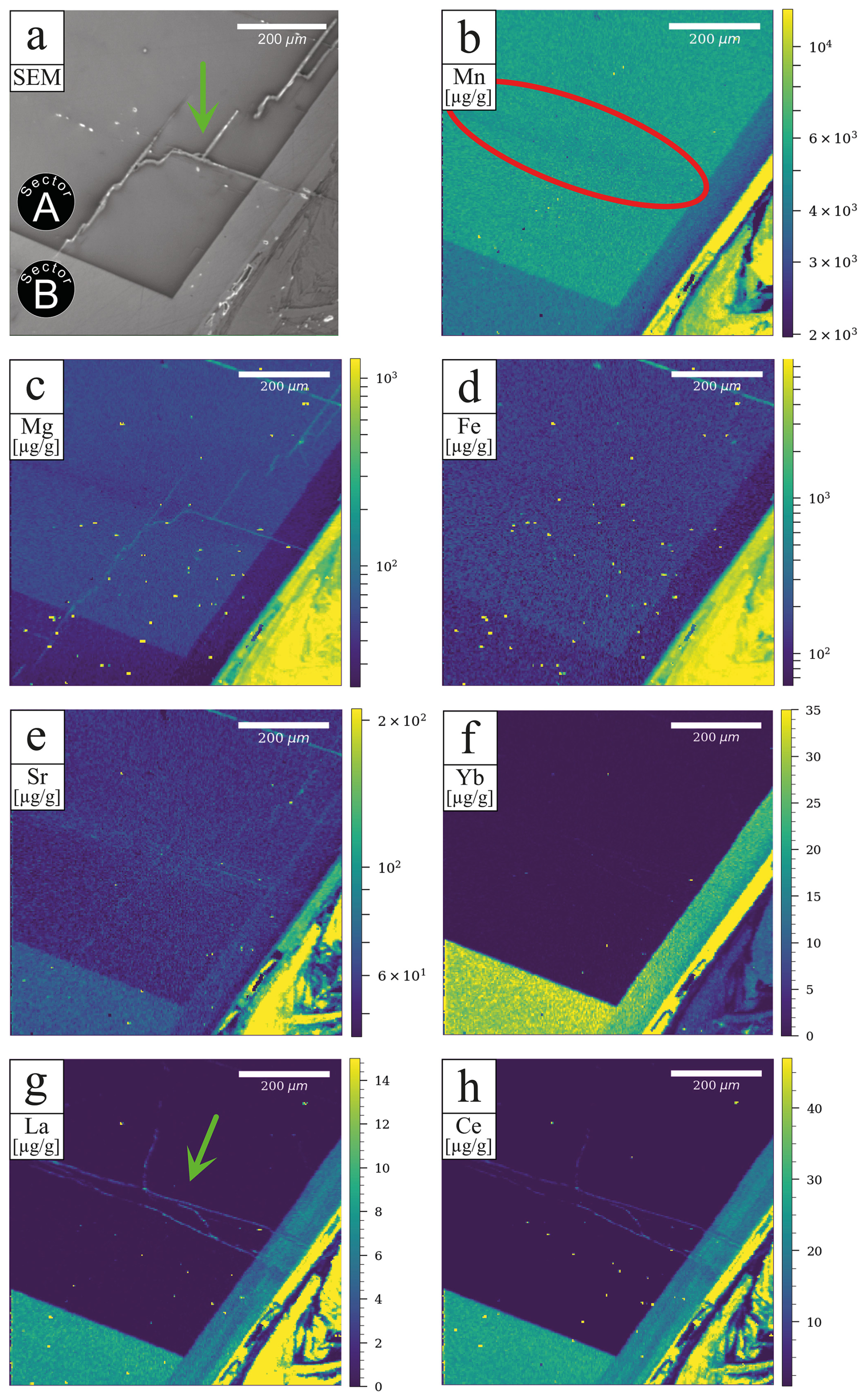
Figure 2SEM image and LA-ICP-MS element maps of the area marked in Fig. 1. A preference of Mn, Mg, and Fe for sector A is striking, contrarily to the preference of Sr, Yb, La, and Ce for sector B. The red area in (b) highlights local depletions associated with linear structures visible in either the SEM image or the element map marked by green arrows in (a) and (g).
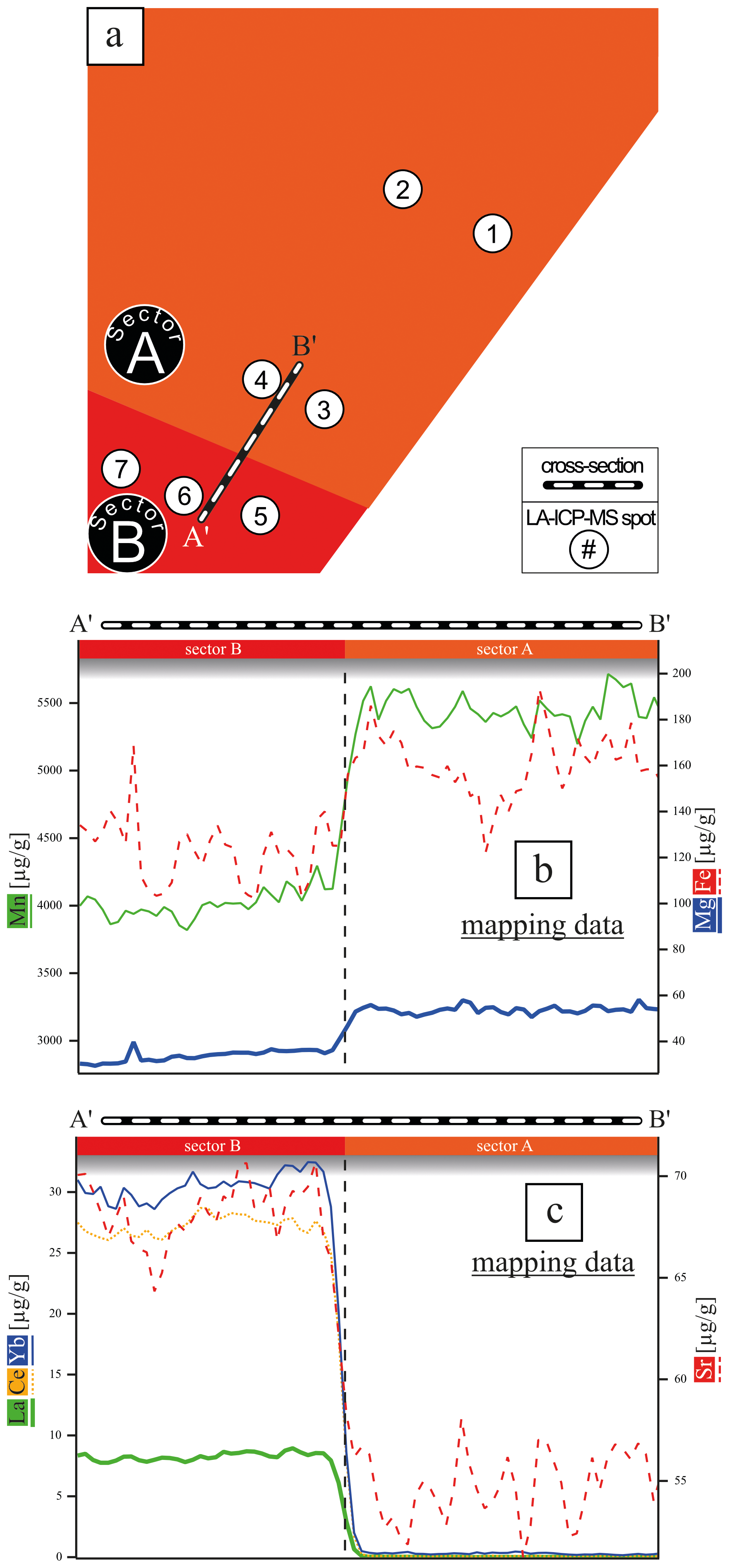
Figure 3(a) Schematic image of the analyzed area with marked cross-section A′B′ covering the sectors A and B. LA-ICP-MS spot analyses with a spot size of 50 µm are marked. (b, c) Element concentration of cations with an affinity for sector A and B. The data were extracted from the LA-ICP-MS element map concentration data (Fig. 2).
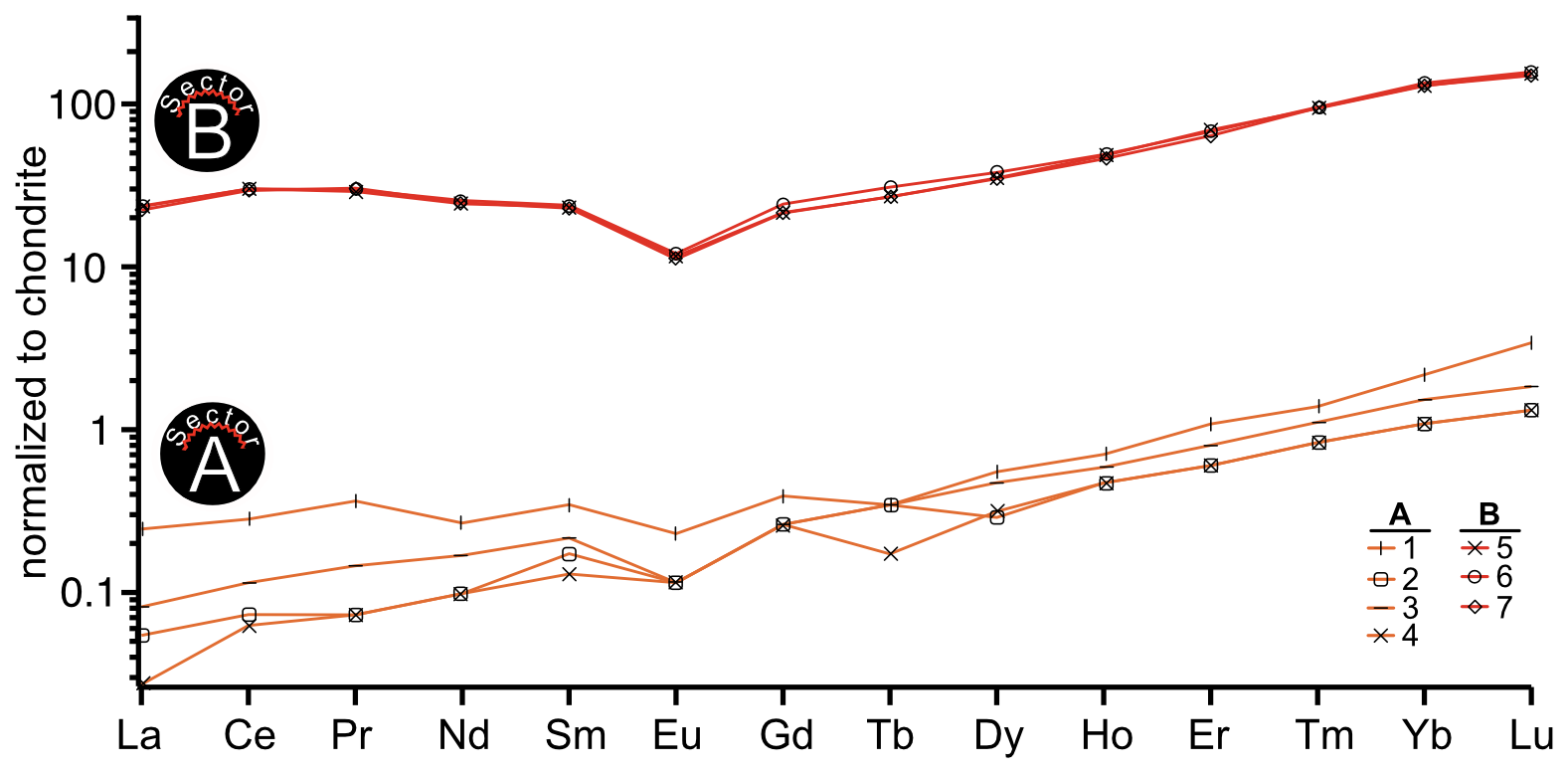
Figure 4REE curve range of LA-ICP-MS spot concentration data. Orange curves indicate spots on sector A, and red curves indicate spots on sector B.
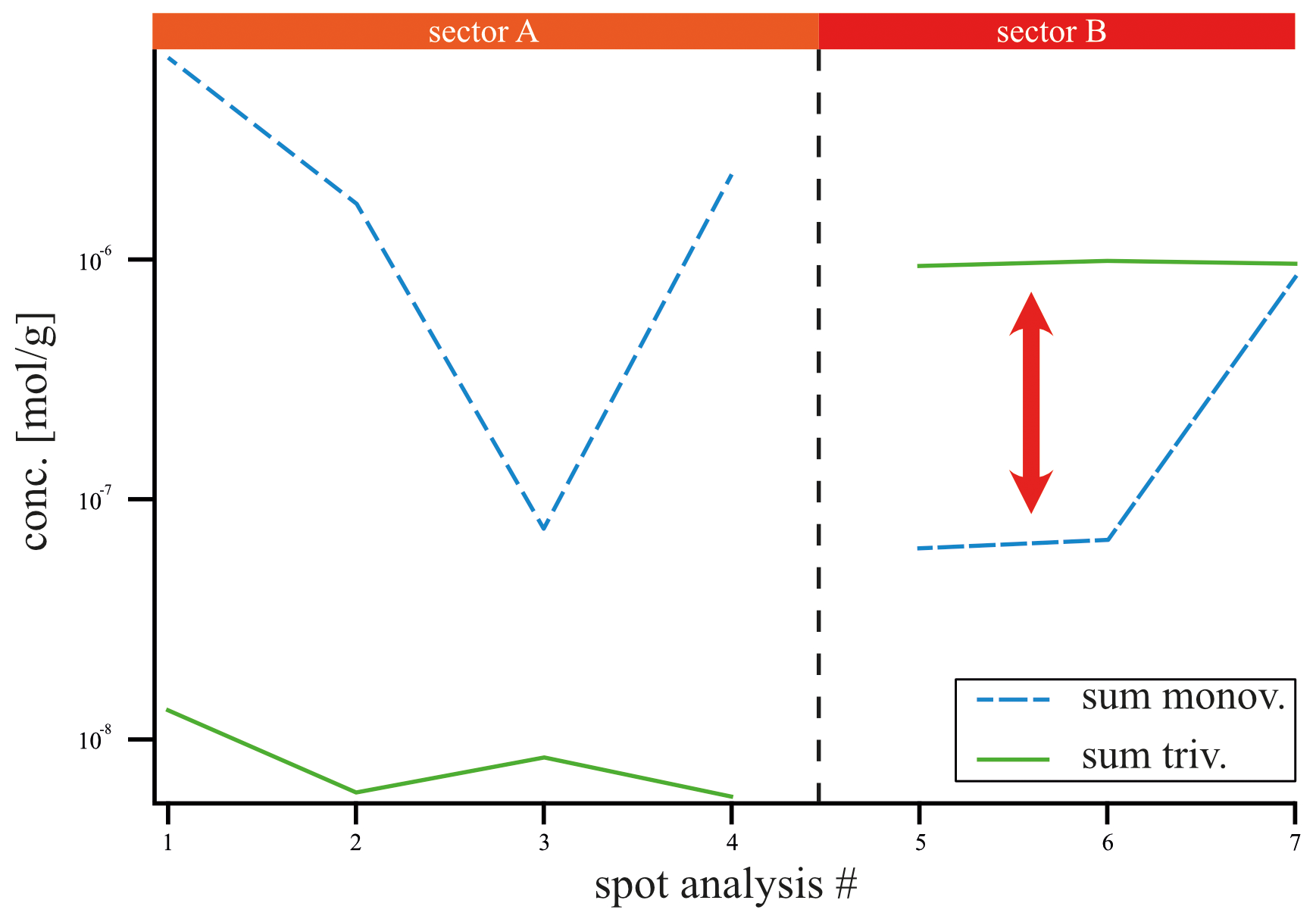
Figure 5Averaged LA-ICP-MS spot concentration data of all monovalent cations (Li, Na, K, Rb; blue) and all REE3+ (green) in mol g−1 over sectors A (spot 1, 2, 3, 4) and B (spot 5, 6, 7). Monovalent and trivalent cations do not follow the same trend across the sectors, which does not support coupled substitution as the driving incorporation mechanism.
Given that the REE exhibited a preferential incorporation into sector B, a similar behavior in sector B is anticipated for the monovalent cations (Li, Na, K, Rb) if coupled substitution occurs. Figure 5 presents the concentrations of these elements, categorized according to the sectors in which the LA-ICP-MS spots were located. In the following, concentrations are expressed in mol g−1 to facilitate the evaluation of coupled substitution, which is governed by the relative numbers (and charge balance) of substituent ions rather than their mass-based concentrations. The Li concentration ranges from 1.44 ± to 5.76 ± mol g−1. With a factor of 1, the incorporation of Li is equally distributed between sectors A and B (Table 1). Sodium concentrations exhibit significant variation, ranging from 4.57 ± to 6.86 ± mol g−1, with a pronounced preference for sector A, indicated by a factor of 8.67. Potassium concentrations range from mol g−1 to 7.95 ± mol g−1, also demonstrating a preference for sector A, with a factor of 4.74. In contrast, Rb displays the lowest concentrations, ranging from 1.17 ± to 3.51 ± mol g−1, with a preference for sector B, indicated by a factor of 1.52. These results clearly show no preference for monovalent cation incorporation into the REE-dominated sector B.
6.1 Sector-selective cation incorporation in calcite
The selective incorporation of cations into specific crystallographic sectors of calcite has been widely documented (e.g., Reeder and Paquette, 1989; Kusturica et al., 2022), but the observed affinities of specific elements for distinct sectors lack uniformity across studies. For instance, Kusturica et al. (2022) hypothesized that trivalent and divalent cations occupy different crystallographic sectors, making the valency the steering factor of sector selection based on their LA-ICP-MS element analysis in secondary calcite.
The observations of the present study align with the idea of divalent cations such as Mn, Mg, and Fe exhibiting coherent behaviors in terms of sectoral distribution (Reeder and Paquette, 1989; Kusturica et al., 2022), with a notable exception: Sr2+ demonstrates a clear affinity for the REE3+-enriched sector B (Fig. 3c). This observation challenges the hypothesis of valence state being the major determinant of sectoral incorporation and suggests that, while valence may influence the sector selectivity, it is not the only parameter to consider. This complexity is further confirmed by the work of Gabitov et al. (2021), who extended this understanding by evaluating the partition coefficients (KTE= ()()fluid) of trace elements in specific calcite crystal faces, revealing significant heterogeneity within individual crystals and with Mg showing contrary incorporation behavior compared to Sr. Furthermore, Barker and Cox (2011) found a weak negative correlation between Sr and Mn within calcite needles, thus ruling out a universal rule for divalent cations.
The ionic size of substituted cations is another relevant factor influencing the sector-selective incorporation in calcite. With Ca having an ionic radius of 0.1 nm (Shannon, 1976; 6-fold coordination), this study observed ions larger than Ca accumulating in sector B (Sr 0.118 nm, La 0.103 nm, Ce 0.101 nm), while smaller ions favor sector A (Mg 0.072 nm, Mn 0.083 nm, Fe 0.078 nm). This observation is supported by Paquette and Reeder (1995), who investigated intrasectoral zoning, which is defined as compositional differences within a single crystallographic face. They suggested that the ionic radius, relative to the Ca2+ ion, plays a critical role in incorporation along step edges formed during spiral growth on the calcite (104) face. Reeder (1996) observed this ion-size-dependent step selection for Co, Cd, Mg, Mn, Ba, Sr, and Pb, with Zn being a notable exception, possibly due to strong interactions with solvent molecules or specific surface sites. These observations for intrasectoral zoning are based on the different incorporation of ions into obtuse and acute growth steps on the (104) face, which can be extended to the varying crystal lattice parameters of different calcite faces. These other faces exhibit a characteristic atomic density, ion coordination, and charge distribution, which influence their capacity for size-selective ion incorporation.
Beyond valence and size, hydration energy is also relevant as a controlling force for the structural uptake of cations into calcite. Smaller and more highly charged ions form stronger hydration shells, which must be overcome for structural incorporation to occur (Burgos-Cara et al., 2017; Dziadkowiec et al., 2021). This parameter significantly influences the sector-selective behavior as different crystallographic faces provide varying energy barriers for dehydration. The (104) face, characterized by its high crystallographic density of 4.95 nm−2 for calcium and carbonate sites, serves as the primary template for mineral–water interactions and exhibits a well-defined interfacial structure with two ordered layers of water at 0.235 ± 0.005 and 0.324 ± 0.006 nm above calcium sites, creating distinct environments for cation dehydration (Heberling et al., 2014). However, the less abundant faces ((001), (012), and (110)) exhibit different atomic arrangements and variations in coordination and charge distribution and also show higher surface energies and correspondingly higher reactivity (Bruno et al., 2013).
Within this framework, hydration energy provides a mechanistic link between face-specific structure and observed sectoral incorporation. For instance, La3+, with its high hydration energy of −3187 kJ mol−1 (Kepp, 2018), experiences greater difficulty in dehydration and incorporation into stable faces, like the (104) face, compared to Fe2+ (−1928 kJ mol−1; Kepp, 2018), Mn2+ (−1848 kJ mol−1), and Mg2+ (−1918 kJ mol−1), which can more readily access less reactive surface sites. The correlating behavior between the REEs with high hydration energy and Sr2+, with a relatively low hydration energy of −1468 kJ mol−1 (Kepp, 2018) according to sector incorporation, can be attributed to the large ionic radius of Sr2+, which excludes Sr in order to incorporate in high quantities into sector A due to crystallographic reasons. This finding underlines the complexity of multiple parameters influencing the sector-selective incorporation process.
The molecular-level understanding of these processes has been significantly advanced by Heberling et al. (2014), who used surface diffraction measurements to characterize the calcite–water interface at the atomic scale. Their findings reveal that surface carbonate ions relax only slightly from bulk positions with an approx. 4° tilt towards the surface, creating specific incorporation sites that favor different cation sizes and hydration states. These structural parameters are in good agreement with those obtained from synchrotron X-ray scattering studies by Fenter et al. (2000) and Fenter and Sturchio (2004), who report minimal relaxation of surface carbonate groups and a similarly ordered interfacial water structure at calcite (104). This structural refinement provides a mechanistic basis for understanding why certain cations preferentially incorporate into specific crystallographic sectors as the local coordination environment and available space at different surface sites directly influence the energetics of cation dehydration and incorporation.
6.2 REE distribution patterns
The REE distribution patterns of the calcite sectors A and B (Fig. 4) exhibit similar shapes, characterized by a flat LREE and MREE segment and a slight increase towards HREE. The similarity in REE curve shapes across the coequally grown sectors A and B suggests that the REE characteristics of the mineral-forming fluid were reflected in different quantities within both sectors. Despite this similar pattern, absolute concentration levels differ significantly, with a distribution factor for La and Ce (Table 1) exceeding 200 between sectors A and B. Previous studies have documented less pronounced sector-related REE partitioning. Barker and Cox (2011) observed REE partitioning in synthetic calcite, with a factor of 2.5 between two coevally grown sectors, while Kusturica et al. (2022) documented a factor of 10 in natural calcites.
Furthermore, Barker and Cox (2011) reported differences in the shapes of REE distribution curves between sectors, particularly elevated MREE concentrations in one sector, suggesting crystallographic control over REE incorporation. Three possibilities may explain the differing REE patterns observed by Barker and Cox (2011) compared to the consistent REE patterns found across the sectors of this study:
-
First, there is the temporal resolution of growth events. While Barker and Cox (2011) interpreted their investigated pair of sectors as having formed during the same growth event, the non-adjacent position of these sectors on the crystal suggest that this may not be the case. It is possible that these sectors actually formed during separate precipitation events, each with different fluid compositions. This could explain the differences observed in their REE signatures.
-
Second, there is sector pair selection. Calcite exhibits a variety of distinct crystallographic faces (Heberling et al., 2011) which pose a characteristic trace element incorporation behavior. Since this study and previous ones focused investigations based on only two sectors, the combinations of such sector pairs selected from the available calcite faces may differ between studies. The high sectoral REE concentration contrast in this study may reflect the selection of sectors with particularly divergent incorporation potentials. Kusturica et al. (2022) have detected multiple sectors within the same growth event of natural calcite, which supports this theory.
-
Third, there are analytical artifacts. The LA-ICP-MS ablation process penetrates beneath the surface of the sample, potentially incorporating undesired material such as fluid inclusions or material from different growth phases. This issue is particularly relevant for analyses conducted near the outermost rim of crystals, as presented by Barker and Cox (2011). They recognized that variable morphology in the third dimension may have influenced their results, potentially contributing to discrepancies in REE distributions.
Beyond the substantial differences in concentration, the patterns for sectors A and B (Fig. 4) exhibit a similar characteristic distribution, with consistent LREE and MREE trends while indicating a slight increase in HREE concentrations. Morgan and Wandless (1980) mentioned that the formation of REE patterns is linked to the ionic size of those REEs. The LREE ionic radii are closer to the radius of Ca and are therefore more easily incorporated into the crystal lattice compared to HREE. This would result in a preferred uptake of LREE compared to HREE. The observed contrary behavior of REEs in this study (Fig. 4) supports the hypothesis that sorption and complexation act as the driving force for fractionation of LREE and HREE, as previously described by Bau (1991) and Subías and Fernández-Nieto (1995). They suggested that sorption is more closely associated with lower pH values and elevated temperatures, whereas complexation predominates in systems characterized by higher pH and decreasing temperatures. The temperature dependency is consistent with the low-temperate hydrothermal calcite growth event of this studied crystal, as described by Burisch et al. (2018). REE complexation behavior varies significantly with fluid composition and pH conditions in hydrothermal systems. In neutral to alkaline hydrothermal systems, REEs mainly exist in fluids as hydroxyl complexes or other ligand-bearing hydroxyl complexes (Di and Ding, 2024). The formation of hydroxyl-carbonate complexes in alkaline fluids specifically enhances hydrothermal mobilization of different REE groups at different temperature ranges, creating conditions favorable for HREE enrichment in lower-temperature calcite precipitation (Louvel et al., 2022).
The presence of a negative Eu anomaly (Fig. 4) in hydrothermal calcite is predominantly linked to the temperature and redox conditions of the hydrothermal fluids from which the calcite precipitates. Negative Eu anomalies in carbonate minerals can occur under low-temperature conditions (< 250 °C), which reflect the behavior of Eu in the fluid phase (Warmada et al., 2007; Zheng et al., 2023). In hydrothermal systems characterized by reducing conditions, the ratio may favor the incorporation of Eu3+ into the carbonate lattice, potentially reducing the overall Eu concentration and creating a negative anomaly in chondrite-normalized REE patterns (Warmada et al., 2007).
6.3 Incorporation mechanisms
The mechanism of charge equilibration for REE incorporation in calcite has previously been assumed to involve either coupled substitution – NaREE(CO3)2 – or a vacancy associated with two trivalent cations – REE2(CO3)3 – for charge equilibration (Elzinga et al., 2002; Marques Fernandes et al., 2008, Voigt et al., 2017).
However, if coupled substitution was the dominant mechanism for charge equilibration of trivalent cations, one would expect to find at least equivalent quantities of monovalent cations present in sector B. Figure 3d presents the concentrations of REEs and monovalent cations across sectors A and B, revealing that monovalent cations are preferentially associated with sector A, contrarily to the distribution of REEs. Specifically, spots 5 and 6 (sector B, Fig. 5) display concentrations of monovalent cations that are 1 order of magnitude lower than those of REEs (see red arrow). Only spot 7 exhibits similar concentrations of monovalent and trivalent cations, which may be linked to impurities or fluid inclusions.
A comparison of the average molar concentrations in sector B (spots 5, 6, and 7) shows that the analyzed monovalent cations (Li, Na, K, Rb; mol g−1, Fig. 5) are present at much lower levels than the total REE concentration ( mol g−1). This disparity suggests that coupled substitution alone cannot account for charge equilibration in this context.
These results indicate that both coupled substitution and charge equilibration via vacancies contribute to REE incorporation in sector B. It should be noted that Cs was not included in this analysis. However, due to its significantly larger ionic radius (0.167 nm; Shannon, 1976) relative to Ca (0.1 nm), Cs is not expected to affect the conclusions regarding the coupled substitution mechanism.
The results of this study provide new constraints on trace element partitioning associated with sector zoning in hydrothermal calcite and its relevance for radionuclide retention in repository environments. Sector-selective partitioning yields REE concentration differences exceeding 200-fold between adjacent sectors, and an ionic radius relative to Ca2+ emerges as the primary control on sector preference, with larger cations (Sr2+, La3+, Ce3+) being enriched in sector B and smaller cations (Mg2+, Mn2+, Fe2+) favoring sector A; the affinity of Sr2+ for the REE-enriched sector B shows that valence alone does not govern sectoral incorporation.
The data further indicate that REE-enriched sectors incorporate trivalent cations via coupled substitution and likely also through vacancy-involved mechanisms: molar concentration balances show that available monovalent cations are insufficient to fully charge-compensate for the REE inventory in sector B, suggesting an additional contribution from vacancies, although this mechanism remains indirect and should be regarded to be plausible rather than demonstrated. REE patterns (flat LREE–MREE, slight HREE enrichment, negative Eu anomalies) are consistent across sectors and reflect low-temperature hydrothermal complexation at 50–70 °C. Overall, this work shows that sector-zoned calcite can locally develop sectors with markedly enhanced capacity for REEs and, by analogy, An3+ incorporation when growth favors the REE-rich sector, implying that calcite retention capacity depends critically on the abundance and preservation of such sectors.
Analytical data are available in the Supplement.
The supplement related to this article is available online at https://doi.org/10.5194/ejm-38-197-2026-supplement.
FBa, MKu, and TN planned the campaign. FBa and MKu performed the LA-ICP-MS measurements. FBa, MKl, and FBr performed the SEM imaging. FBa analyzed the data. FBa wrote the paper draft. MKu, TN, MKl, and FBr reviewed and edited the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We thank Herbert Matthes, who guided us through the Wenzel mine and supplied us with helpful information. This work was supported by the Federal Ministry of Education and Research (BMBF) joint research project KRIMI “Kinetik der Radionuklidimmobilisierung endlagerrelevanter Mischkristalle” (grant no. 02NUK056D).
During the preparation of this work, the authors used perplexity.ai in order to improve readability and language. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
This research has been supported by the Bundesministerium für Forschung, Technologie und Raumfahrt (grant no. 02NUK056D).
The publication of this article was funded by the Open Access Publication Fund of TU Berlin.
This paper was edited by Encarnacion Ruiz-Agudo and reviewed by Rinat Gabitov and one anonymous referee.
Apted, M. J. and Ahn, J.: Repository 101: Multiple-barrier geological repository design and isolation strategies for safe disposal of radioactive materials, Woodhead Publ. Series Energy, 2017, 3–26, https://doi.org/10.1016/B978-0-08-100642-9.00001-3, 2017.
Baatartsogt, B., Schwinn, G., Wagner, T., Taubald, H., Beitter, T., and Markl, G.: Contrasting paleofluid systems in the continental basement: a fluid inclusion and stable isotope study of hydrothermal vein mineralization, Schwarzwald district, Germany, Geofluids, 7, 123–147, https://doi.org/10.1111/j.1468-8123.2007.00169.x, 2007.
Barker, S. L. L. and Cox, S. F.: Oscillatory zoning and trace element incorporation in hydrothermal minerals: insights from calcite growth experiments, Geofluids, 11, 48–56, https://doi.org/10.1111/j.1468-8123.2010.00305.x, 2011.
Bau, M.: Rare-earth element mobility during hydrothermal and metamorphic fluid-rock interaction and the significance of the oxidation state of europium, Chem. Geol., 93, 219–230, https://doi.org/10.1016/0009-2541(91)90115-8, 1991.
Birkholzer, J., Houseworth, J., and Tsang, C. F.: Geologic disposal of high-level radioactive waste: status, key issues, and trends, Annu. Rev. Environ. Resour., 37, 79–106, https://doi.org/10.1146/annurev-environ-090611-143314, 2012.
Bruno, J. and Sandino, A.: Radionuclide co-precipitation, SKB Tech. Rep., 87-23, 1987.
Bruno, M., Massaro, F. R., Pastero, L., Costa, E., Rubbo, M., Prencipe, M., and Aquilano, D.: New estimates of the free energy of calcite/water interfaces for evaluating the equilibrium shape and nucleation mechanisms, Cryst. Growth Des., 13, 1170–1179, https://doi.org/10.1021/cg3015817, 2013.
Burgos-Cara, A., Putnis, C. V., Rodriguez-Navarro, C., and Ruiz-Agudo, E.: Hydration effects on the stability of calcium carbonate pre-nucleation species, Minerals, 7, 126, https://doi.org/10.3390/min7070126, 2017.
Burisch, M., Walter, B. F., Gerdes, A., Lanz, M., and Markl, G.: Late-stage anhydrite-gypsum-siderite-dolomite-calcite assemblages record the transition from a deep to a shallow hydrothermal system in the Schwarzwald mining district, SW Germany, Geochim. Cosmochim. Ac., 223, 259–278, https://doi.org/10.1016/j.gca.2017.12.002, 2018.
Curti, E.: Coprecipitation of radionuclides with calcite: estimation of partition coefficients based on a review of laboratory investigations and geochemical data, Appl. Geochem., 14, 433–445, https://doi.org/10.1016/S0883-2927(98)00065-1, 1999.
Di, J. and Ding, X.: Complexation of REE in hydrothermal fluids and its significance on REE mineralization, Minerals, 14, 531, https://doi.org/10.3390/min14060531, 2024.
Drake, H., Mathurin, F. A., Zack, T., and Schäfer, T.: Incorporation of trace elements into calcite precipitated from deep anoxic groundwater in fractured granitoid rocks, Procedia Earth Planet. Sci., 17, 841–844, https://doi.org/10.1016/j.proeps.2017.01.056, 2017.
Dziadkowiec, J., Ban, M., Javadi, S., Jamtveit, B., and Royne, A.: Ca2+ ions decrease adhesion between two (104) calcite surfaces as probed by atomic force microscopy, ACS Earth Space Chem., 5, 2827–2838, https://doi.org/10.1021/acsearthspacechem.1c00220, 2021.
Elzinga, E. J., Reeder, R. J., Withers, S. H., Peale, R. E., Manson, R. A., Beck, K. M., and Hess, W. P.: EXAFS study of rare-earth element coordination in calcite, Geochim. Cosmochim. Ac., 66, 2875–2885, https://doi.org/10.1016/S0016-7037(02)00888-8, 2002.
Fenter, P. and Sturchio, N. C.: Mineral-water interfacial structures revealed by synchrotron X-ray scattering, Prog. Surf. Sci., 77, 171–258, https://doi.org/10.1016/j.progsurf.2004.12.001, 2004.
Fenter, P., Geissbühler, P., DiMasi, E., Srajer, G., Sorensen, L. B., and Sturchio, N. C.: Surface speciation of calcite observed in situ by high-resolution X-ray reflectivity, Geochim. Cosmochim. Ac., 64, 1221–1228, https://doi.org/10.1016/S0016-7037(99)00403-2, 2000.
Gabitov, R. I., Sadekov, A., and Migdisov, A.: REE incorporation into calcite individual crystals as one time spike addition, Minerals, 7, 204, https://doi.org/10.3390/min7110204, 2017.
Gabitov, R. I., Sadekov, A., Dyer, J., Perez-Huerta, A., Xu, H., and Migdisov, A.: Sectoral and growth rate control on elemental uptake by individual calcite crystals, Chem. Geol., 585, 120589, https://doi.org/10.1016/j.chemgeo.2021.120589, 2021.
Geyer, O. F., Gwinner, M. P., Geyer, M., Nitsch, E., and Simon, T.: Geologie von Baden-Württemberg, Schweizerbart, Stuttgart, 627 pp., ISBN 978-3-510-65267-9, 2011.
Hann, H. P.: Tektonik und Petrologie des Südschwarzwälder Kristallins im Gebiet des unteren Wehratals, Neues Jahrb. Geol. Paläontol. Abh., 240, 121–151, https://doi.org/10.1127/njgpa/240/2006/121, 2006.
Heberling, F., Trainor, T. P., Lützenkirchen, J., Eng, P., Denecke, M. A., and Bosbach, D.: Structure and reactivity of the calcite-water interface, J. Colloid Interf. Sci., 354, 843–857, https://doi.org/10.1016/j.jcis.2010.10.047, 2011.
Heberling, F., Bosbach, D., Eckhardt, J. D., Fischer, U., Glowacky, J., Haist, M., Kramar, U., Loos, S., Müller, H. S., Neumann, T., Pust, C., Schäfer, T., Stelling, J., Ukrainczyk, M., Vinograd, V., Vucak, M., and Winkler, B.: Reactivity of the calcite-water interface, from molecular scale processes to geochemical engineering, Appl. Geochem., 45, 158–190, https://doi.org/10.1016/j.apgeochem.2014.03.006, 2014.
Heller, A., Barkleit, A., Foerstendorf, H., Tsushima, S., Heim, K., and Bernhard, G.: Curium(III) citrate speciation in biological systems: a europium(III)-assisted spectroscopic and quantum chemical study, Dalton Trans., 41, 13969, https://doi.org/10.1039/c2dt31480k, 2012.
Heller, A., Senwitz, C., Foerstendorf, H., Tsushima, S., Holtmann, L., Drobot, B., and Kretzschmar, J.: Europium(III) meets etidronic acid (HEDP): a coordination study combining spectroscopic, spectrometric, and quantum chemical methods, Molecules, 28, 4469, https://doi.org/10.3390/molecules28114469, 2023.
Jensen, M. P. and Bond, A. H.: Comparison of covalency in the complexes of trivalent actinide and lanthanide cations, J. Am. Chem. Soc., 124, 9870–9877, https://doi.org/10.1021/ja0178620, 2002.
Kalt, A., Altherr, R., and Hanel, M.: Contrasting P–T conditions recorded in ultramafic high-pressure rocks from the Variscan Schwarzwald (F.R.G.), Contrib. Mineral. Petrol., 121, 45–60, https://doi.org/10.1007/s004100050089, 1995.
Kalt, A., Altherr, R., and Hanel, M.: The Variscan basement of the Schwarzwald, Ber. Dtsch. Mineral. Ges., 1–43, https://www.researchgate.net/publication/269092883_The_Variscan_Basement_of_the_Schwarzwald (last access: 17 April 2026), 2000.
Kepp, K. P.: Thermochemically consistent free energies of hydration for di- and trivalent metal ions, J. Phys. Chem. A, 122, 7464–7471, https://doi.org/10.1021/acs.jpca.8b06674, 2018.
Kim, J. S., Kwon, S. K., Sanchez, M., and Cho, G. C.: Geological storage of high-level nuclear waste, KSCE J. Civ. Eng., 15, 721–737, https://doi.org/10.1007/s12205-011-0012-8, 2011.
Krauskopf, K. B.: Thorium and rare-earth metals as analogs for actinide elements, Chem. Geol., 55, 323–335, https://doi.org/10.1016/0009-2541(86)90033-1, 1986.
Kurniawan, T. A., Othman, M. H. D., Singh, D., Avtar, R., Hwang, G. H., Setiadi, T., and Lo, W.-H.: Technological solutions for long-term storage of partially used nuclear waste: a critical review, Ann. Nucl. Energy, 166, 108736, https://doi.org/10.1016/j.anucene.2021.108736, 2021.
Kusturica, A., van Loon, N., Drake, H., and Schäfer, T.: LA-ICP-MS analysis of trace and rare-earth element distribution in calcite fracture fillings from Forsmark, Simpevarp and Laxemar (Sweden), Environ. Earth Sci., 81, 371, https://doi.org/10.1007/s12665-022-10462-1, 2022.
Lakshtanov, L. Z. and Stipp, S. L. S.: Experimental study of europium(III) coprecipitation with calcite, Geochim. Cosmochim. Ac., 68, 819–827, https://doi.org/10.1016/j.gca.2003.07.010, 2004.
Louvel, M., Etschmann, B., Guan, Q., Testemale, D., and Brugger, J.: Carbonate complexation enhances hydrothermal transport of rare earth elements in alkaline fluids, Nat. Commun., 13, 1456, https://doi.org/10.1038/s41467-022-28943-z, 2022.
Marques Fernandes, M., Schmidt, M., Stumpf, T., Walther, C., Bosbach, D., Klenze, R., and Fanghänel, T.: Site-selective time-resolved laser fluorescence spectroscopy of Eu3+ in calcite, J. Colloid Interf. Sci., 321, 323–331, https://doi.org/10.1016/j.jcis.2008.01.017, 2008.
Metz, R., Richter, M., and Schürenberg, H.: Die Blei-Zink-Erzgänge des Schwarzwaldes, Amt für Bodenforschung, Beihefte zum Geologischen Jahrbuch, Stuttgart, Beiheft 29, 277 pp., ES184032900, http://www.schweizerbart.de//publications/detail/artno/184032900/Beih_29_z_Geol_Jahrbuch_vergriffen (last access: 17 April 2026), 1957.
Möller, P. and De Lucia, M.: Incorporation of rare earths and yttrium in calcite: a critical re-evaluation, Aquat. Geochem., 26, 89–117, https://doi.org/10.1007/s10498-020-09369-9, 2020.
Morgan, J. W. and Wandless, G. A.: Rare earth element distribution in some hydrothermal minerals: evidence for crystallographic control, Geochim. Cosmochim. Ac., 44, 973–980, https://doi.org/10.1016/0016-7037(80)90286-0, 1980.
Nedel, S., Didieriksen, K., Christiansen, B. C., Bovet, N., and Stipp, S. L. S.: Uptake and release of cerium during Fe-oxide formation and transformation in Fe(II) solutions, Environ. Sci. Technol., 44, 4493–4498, https://doi.org/10.1021/es9031503, 2010.
Olszewska, A., Miskiewicz, A., Zakrzeska-Koltuniewicz, G., Lankof, L., and Pajak, L.: Multi-barrier system against migration of radionuclides from radioactive waste repository, Nukleonika, 60, 557–563, https://doi.org/10.1515/nuka-2015-0103, 2015.
Paquette, J. and Reeder, R. J.: Relationship between surface structure, growth mechanism, and trace element incorporation in calcite, Geochim. Cosmochim. Ac., 59, 735–749, https://doi.org/10.1016/0016-7037(95)00004-J, 1995.
Perry, E. P.: Rare earth element signatures in hydrothermal calcite: insights from numerical modeling, experimental geochemistry and mineral deposits in New Mexico, PhD thesis, Colorado School of Mines, 13857310, https://doi.org/10.1155/2018/5382480, 2019.
Pflug, R.: Bau und Entwicklung des Oberrheingrabens, Wiss. Buchges., Darmstadt, 145 pp., ISBN 3-534-07186-7, 1982.
Reeder, R. J. and Paquette, J.: Sector zoning in natural and synthetic calcites, Sediment. Geol., 65, 239–247, https://doi.org/10.1016/0037-0738(89)90026-2, 1989.
Reeder, R. J.: Interaction of divalent cobalt, zinc, cadmium, and barium with the calcite surface during layer growth, Geochim. Cosmochim. Ac., 60, 1543–1552, https://doi.org/10.1016/0016-7037(96)00034-8, 1996.
Schwinn, G. and Markl, G.: REE systematics in hydrothermal fluorite, Chem. Geol., 216, 225–248, https://doi.org/10.1016/j.chemgeo.2004.11.012, 2005.
Shannon, R. D.: Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr. A, 32, 751–767, https://doi.org/10.1107/S0567739476001551, 1976.
Staude, S., Mordhorst, T., Nau, S., Pfaff, K., Brügmann, G., Jacob, D. E., and Markl, G.: Hydrothermal carbonates of the Schwarzwald ore district, southwestern Germany: carbon source and conditions of formation using δ18O, δ13C, 87Sr/86Sr, and fluid inclusions, Can. Mineral., 50, 1401–1434, https://doi.org/10.3749/canmin.50.5.1401, 2012.
Stipp, S. L. S., Christensen, J. T., Lakshtanov, L. Z., Baker, J. A., and Waight, T. E.: Rare earth element (REE) incorporation in natural calcite: upper limits for actinide uptake in a secondary phase, Radiochim. Acta, 94, 523–528, https://doi.org/10.1524/ract.2006.94.9-11.523, 2009.
Subías, I. and Fernández-Nieto, C.: Hydrothermal events in the Valle de Tena (Spanish Western Pyrenees) as evidenced by fluid inclusions and trace-element distribution from fluorite deposits, Chem. Geol., 124, 267–282, https://doi.org/10.1016/0009-2541(95)00060-Y, 1995.
Voigt, M., Mavromatis, V., and Oelkers, E. H.: The experimental determination of REE partition coefficients in the water-calcite system, Chem. Geol., 462, 30–43, https://doi.org/10.1016/j.chemgeo.2017.04.024, 2017.
Walter, B. F., Immenhauser, A., Geske, A., and Markl, G.: Exploration of hydrothermal carbonate magnesium isotope signatures as tracers for continental fluid aquifers, Schwarzwald mining district, SW Germany, Chem. Geol., 400, 87–105, https://doi.org/10.1016/j.chemgeo.2015.02.009, 2015.
Warmada, W., Lehmann, B., Simandjuntak, M., and Hemes, H. S.: Fluid inclusion, rare-earth element and stable isotope study of carbonate minerals from the Pongkor epithermal gold-silver deposit, West Java, Indonesia, Resour. Geol., 57, 124–135, https://doi.org/10.1111/j.1751-3928.2007.000012.x, 2007.
Werner, W. and Dennert, V.: Lagerstätten und Bergbau im Schwarzwald, Landesamt Geol. Rohst. Bergbau Baden-Württemberg, ISBN-10 3000146369, 2004.
Xue, Y., Sun, D., Wang, L., and Xu, Y.: A double-layered model for near-field temperature in a nuclear waste repository, Prog. Nucl. Energy, 133, 103646, https://doi.org/10.1016/j.pnucene.2021.103646, 2021.
Zhao, H. G., Shao, H., Kunz, H., Wang, J., Su, R., and Liu, Y. M.: Numerical analysis of thermal process in the near field around vertical disposal of high-level radioactive waste, J. Rock Mech. Geotech. Eng., 6, 55–60, https://doi.org/10.1016/j.jrmge.2013.09.004, 2014.
Zheng, J., Wen, W., Ge, Y., Zhou, G., Zhang, Y., Yan, W., Jiang, H., Zhang, Z., and Xi, A.: U–Pb dating of fibrous dolomite in the hydrothermal dolostone of the Dengying Formation, central Sichuan Basin, and its response to supercontinent breakup, Minerals, 13, 1353, https://doi.org/10.3390/min13101353, 2023.
Zhong, S. and Mucci, A.: Partitioning of rare earth elements (REEs) between calcite and seawater solutions at 25 °C and 1 atm, and high dissolved REE concentrations, Geochim. Cosmochim. Ac., 59, 443–453, https://doi.org/10.1016/0016-7037(94)00381-U, 1995.