the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Jan 2025
| 17 Jan 2025
A reliable analytical procedure to determine the carbon isotopic signature of CO2-bearing COH fluids generated in petrological experiments
Luca Minopoli
Elena Ferrari
Simone Tumiati
The ratio of stable carbon isotopes, δ13C, serves as a fundamental tracer for geological processes. Experiments aiming to replicate isotopic exchange between carbon reservoirs encounter significant analytical challenges due to the limited sample size and issues related to sampling, particularly when dealing with volatile species. Here we present a novel methodology that integrates a capsule-piercing device, a quadrupole mass spectrometer (QMS), and isotopic ratio mass spectrometry (IRMS) to measure the CO2 concentration and natural-like δ13C ratio of CO2 in the volatile COH phase generated in petrological experiments. To validate the technique, we first analyze the COH fluid resulting from the thermal decomposition of 1 mg of anhydrous oxalic acid. The optimal values of the carrier gas flow in the QMS, sampling times, and chromatography column temperature for IRMS are determined. The high degree of similarity, within acceptable errors, observed in both compositional and isotopic analyses indicates a robust reproducibility, minimally affected by contamination and fractionation effects during sampling. We also show that this methodology can be applied for estimating the δ13C of CO2 produced from high-pressure, high-temperature, redox-buffered piston–cylinder experiments. This offers a multitude of opportunities in designing experiments focused on determining isotopic fractionation models for geological processes that involve, but are not restricted to, CO2-bearing COH fluids.
- Article
(2384 KB) - Full-text XML
-
Supplement
(1722 KB) - BibTeX
- EndNote





Fluids produced in high-pressure, high-temperature piston–cylinder petrological experiments are contained in capsules and are commonly analyzed ex situ. After quenching, the capsules are removed from the experimental apparatus and opened; then, the fluids are sampled and analyzed using either gas chromatography or mass spectrometry (e.g., Eggler et al., 1979; Rosenbaum and Slagel, 1995). For the analysis by mass spectrometry, Tiraboschi et al. (2016) designed a capsule-piercing device connected to a quadrupole mass spectrometer (QMS; Fig. 1) to quantify the bulk C, O, and H (N) composition of experimental fluids by measuring the volatile species H2O, CO2, CH4, CO, H2, O2, and N2. Due to a high sensitivity and precision – for most of the species the detection limits are less than 1 µmol and the uncertainty in the measurement is on the order of 1 % – this technique has proven to be a valuable tool in evaluating the accuracy of thermodynamic models that predict the speciation of COH fluids and assessing some important deviations (Miozzi and Tumiati, 2020; Peng et al., 2022; Tiraboschi et al., 2018, 2022; Toffolo et al., 2023; Tumiati et al., 2017, 2020). By scanning specific mass-to-charge () ratios the QMS can also detect the isotopic variants of the above species (Tumiati et al., 2022). Nevertheless, the low natural abundances of the common stable isotopes of C, O, and H coupled with the scarce overall mass of the volatile phase in the capsule (typically less than 100 µmol; e.g., Tiraboschi et al., 2016; Toffolo et al., 2023; Tumiati et al., 2020) translates into signals that are too weak, hampering an accurate quantification. This hindrance can be bypassed by designing high-pressure, high-temperature experiments in which isotopically doped materials are used. For instance, Tumiati et al. (2022) applied this approach to monitor the δ13C of CO2 produced under subduction conditions (3 GPa, 700 °C) from the interaction between water and a model sediment composed of synthetic graphite and labeled 13C-rich CaCO3. The results were used to determine that the δ13C (expressed as difference to the Vienna Pee Dee Belemnite (VPDB), international reference standard) of volcanic arc emissions is possibly controlled by CO2 produced through the irreversible oxidative dissolution of organic matter (graphite) and its isotopic buffering with carbonate sediments (CaCO3). However, in circumstances where the isotopic characterization of natural materials is necessary, the best analytical method is to use isotopic ratio mass spectrometry (IRMS), which has been developed to be sensitive to naturally occurring variations in stable isotopes (as low as ∼ 0.1 ‰) of light elements (H, C, O, N, S) (Brand et al., 2015; Meier-Augenstein, 1999). As an example, Kueter et al. (2019a) used IRMS to measure the δ13C of coexisting CH4, CO, CO2, and C produced by high-temperature (300–1200 °C), near-atmospheric-pressure pyrolysis of organic compounds characterized by natural isotopic abundances; the results were used to obtain a calibration of CO2–CH4, CO2–CO, and CH4–CO carbon isotope fractionation factors.
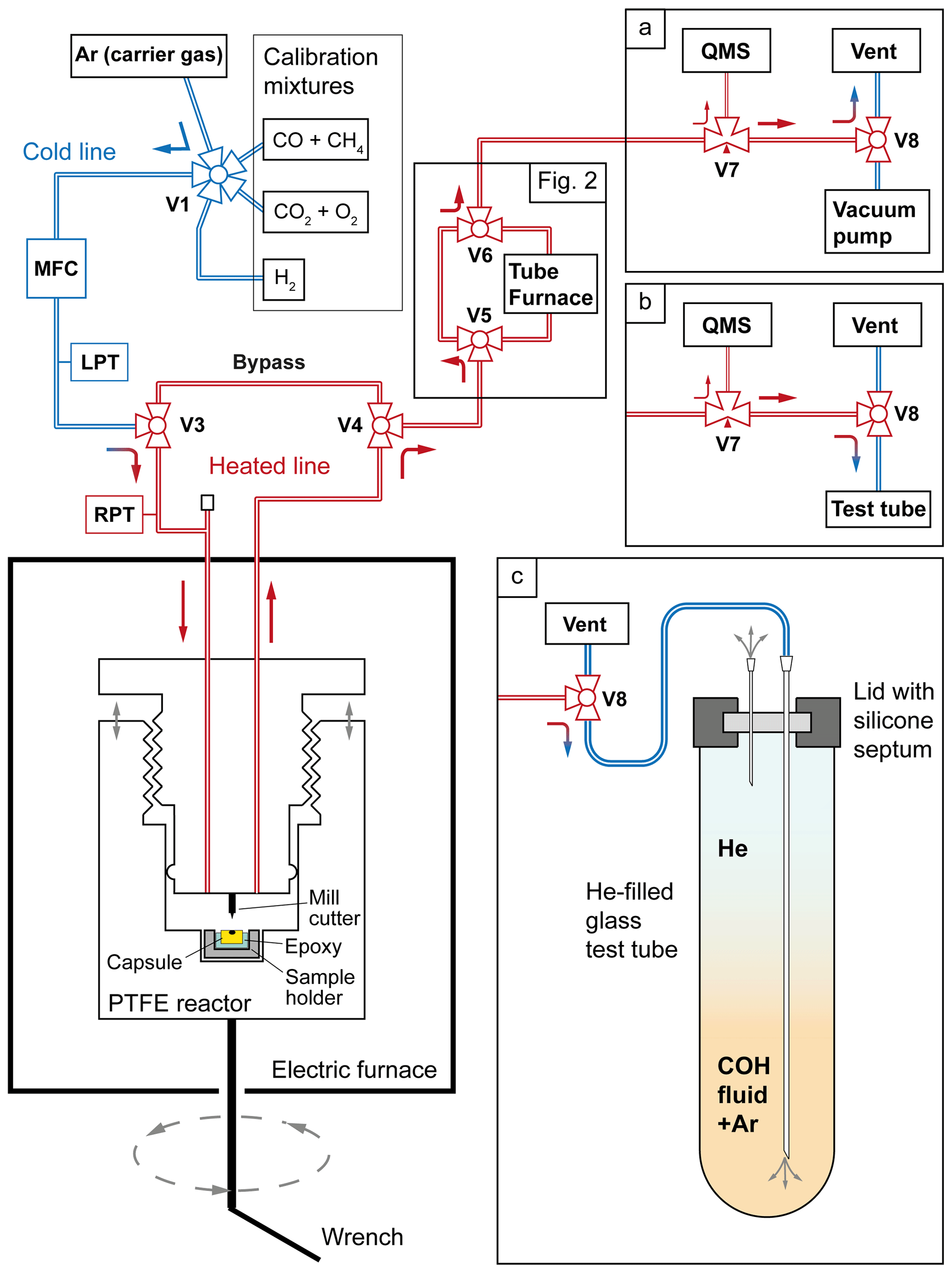
Figure 1Layout of the line connecting the capsule-piercing device with the QMS (adapted from Tiraboschi et al., 2016). In (a) the standard analytical setup is shown, where most of the COH fluid is vented into the atmosphere (“Vent”). In (b) the vacuum pump is disconnected in order to convey the volatile phase to a test tube. In (c) a detailed view of the sampling process, where the gas flows through a syringe needle into a test tube pre-filled with He. MFC: mass flow controller; LPT: line pressure transducer; RPT: reactor pressure transducer.
In this article we provide a new methodology that combines the capsule-piercing device with QMS and IRMS in order to determine both the composition and the ratio of stable carbon isotopes, δ13C, of experimentally produced COH fluids. We took advantage of the fact that in the apparatus of Tiraboschi et al. (2016) only a small fraction of the volatile phase is actually conveyed to the QMS; the rest is vented to the atmosphere. Here, we designed a sampling strategy and tuned an analytical protocol to let the IRMS analyze the volume of the gas that is usually discarded. We demonstrated that this method can be successfully used to measure the δ13C of CO2 with a natural isotopic signature generated in petrological experiments. This will open new possibilities to study the carbon cycle in crustal and mantle systems, where understanding the fractionation of stable carbon isotopes is essential for deciphering the origin and evolution of rocks and fluids (e.g., Luque et al., 2012; Mason et al., 2017; Stachel et al., 2017; Tumiati et al., 2022).
2.1 Experimental strategy
In high-temperature, high-pressure experiments, samples are often contained within sealed noble metal capsules. At our laboratory (Experimental Petrology Laboratory, DiSTAD, University of Milan), we commonly use two experimental capsule configurations for experiments involving a volatile phase.
-
Single capsule. This setup uses capsules with a maximum inner diameter of 4.3 mm and a length of ∼ 10 mm, providing a sample volume of up to ∼ 70 mm3.
-
Double capsule. This configuration is used when redox buffering is required (Eugster, 1957; Eugster and Skippen, 1967). The inner capsule, with a maximum inner diameter of 2 mm and a length of ∼ 8 mm, provides a sample volume of up to ∼ 25 mm3.
Typically, the total amount of volatiles measured after the run is less than 100 µmol (see Tiraboschi et al., 2016; Toffolo et al., 2023; Tumiati et al., 2020). Based on our experience, with the above capsule dimensions, larger quantities of volatiles increase the risk of capsule explosion during the experiment or during quench and depressurization.
Given the small amount of volatiles involved, our primary goal was to evaluate the feasibility and reproducibility of δ13C determination for CO2. To achieve this, we conducted a series of experiments in which a COH fluid was generated through the decomposition of anhydrous oxalic acid (C2H2O4; OAA) – which is a potential source of COH fluids in petrological experiments (e.g., Tiraboschi et al., 2018) – at 250 °C and near-ambient pressures. We then explored potential applications of this method and compared our results with isotopic fractionation models to evaluate the attainment of equilibrium in redox-buffered, high-pressure, high-temperature experiments. Specifically, we performed double-capsule experiments, where COH fluid was obtained from the pyrolysis of organic matter (green microalgae), simulating sub-arc subduction conditions at 3 GPa and 700 °C and with oxygen fugacity (fO2) buffered to ΔFMQ (i.e., 0.8 log units higher than the fO2 imposed by the assemblage fayalite–magnetite–quartz; see Sect. 2.1.2).
2.1.1 Anhydrous oxalic acid (OAA) decomposition experiments
In the first set of experiments (OAA_1, OAA_2, OAA_3, OAA_4; Table 1), a small quantity (1 mg) of pure OAA (≥ 99.0 %; Sigma-Aldrich®; δ13C; see Sect. 2.2.4) was inserted into single gold capsules (outer diameter: 4.5 mm; inner diameter: 4.3 mm; length: 10 mm), which were consequently sealed using a pulse arc welder. In one experiment (OAA_4) the capsule also contained a fragment of a Pt-plated Ni foam catalyst (manufactured by Recemat BV). The capsules were maintained in an oven at 250 °C for an hour in order to attain the complete decomposition of the OAA according to Reaction (1).
Reaction (1) is reported to not produce any solid residue, which could potentially fractionate a part of the carbon (Kueter et al., 2019a; Tiraboschi et al., 2018). The decomposition of 1 mg of OAA will produce a maximum of ∼ 68 µmol of total volatile species, therefore being comparable to the content measured in typical petrological experiments.
A blank experiment, necessary to assess the potential contribution to the δ13C of CO2 from contaminant air, was also performed by sealing an empty gold capsule and heating it for 1 h at 250 °C.
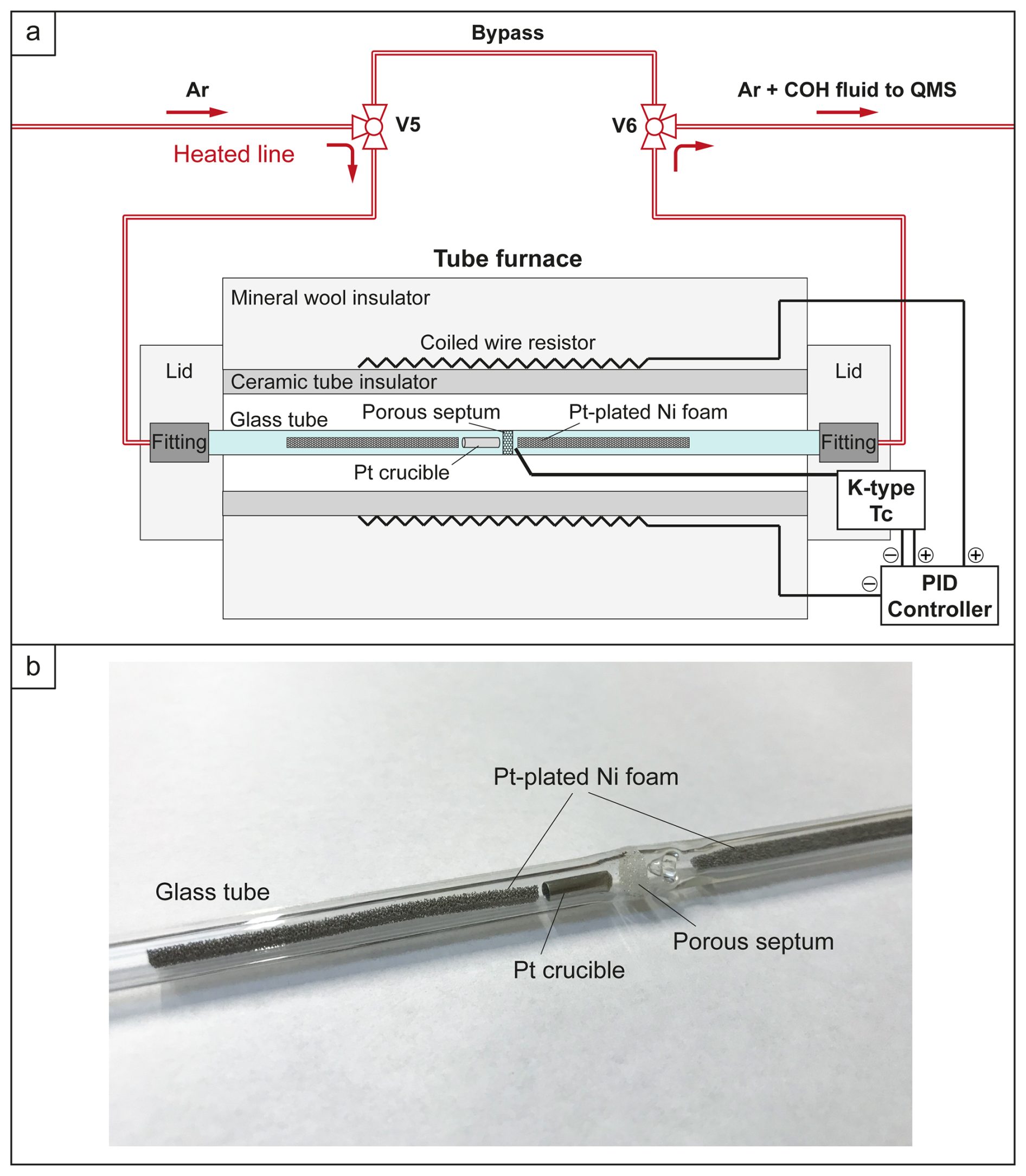
Figure 2The horizontal tube furnace used for OAA decomposition test. (a) Schematic section of the furnace in operation: the temperature in the furnace is monitored through a K-type thermocouple (Tc) and maintained through a PID controller acting on the resistor (heater); Ultra-Torr fittings provide gas-tight sealing. (b) Detail of the borosilicate glass tube containing the Pt crucible and two Pt-plated Ni foam catalysts.
An additional experiment (OAA_5) was conducted to decompose OAA at 250 °C directly within the QMS line, allowing an independent determination of the chemical and isotopic signature of the volatile phase produced. This approach also minimizes potential mass loss that might occur during capsule preparation (e.g., from heating the OAA during welding) or during the experimental run (e.g., H2 loss through the metal walls of the capsule). In this experiment, 1 mg of OAA was placed in a Pt crucible, which was then loaded into a Colaver® borosilicate glass tube (outer diameter: 7 mm; inner diameter: 4 mm; length: 192 mm) along with two bars of Pt-plated Ni foam catalyst (Fig. 2). The glass tube, provided with a porous septum in its middle section, was positioned within a tube furnace and connected to the heated QMS line via two gas-tight Swagelok® Ultra-Torr fittings. Two three-way valves (V5 and V6 in Figs. 1 and 2) allowed the heated QMS line to be diverted to the furnace. The decomposition of OAA was performed by isolating the furnace from the QMS line, heating the sample to 250 °C, and holding this temperature for 1 h. The furnace temperature was monitored and controlled using a K-type thermocouple connected to an Eurotherm® PID (proportional–integral–derivative) controller.
2.1.2 Organic matter pyrolysis experiments
Two redox-buffered high-pressure and high-temperature experiments (COH210, COH245; Table 1) were conducted using commercially available green microalgae Tetraselmis suecica (δ13C; Corg∼ 25 wt %; see Sect. 2.2.4) as a source of carbon. The choice of using green microalgae instead of graphite stems from the fact that the pyrolysis of organic matter produces char – a form of disordered carbonaceous material – which is expected to be more reactive than crystalline graphite (Toffolo et al., 2023; Tumiati et al., 2020). This increased reactivity enhances the likelihood of reaching chemical equilibrium with the COH fluid within the limited time frame of the experimental run, thereby also increasing the potential for achieving isotopic equilibrium (Horita, 2001). The algae were preliminarily washed and centrifuged to remove any water-soluble salt, then dried at 70 °C, and inserted into an Au60Pd40 alloy capsule (outer diameter: 2.3 mm; inner diameter: 2 mm; length: 7–8 mm). In experiment COH245, ∼ 1 µL of ultrapure water (Milli-Q®, treated using nitrogen stripping, while boiling to remove dissolved atmospheric CO2) was added to the algae to maximize CO2 production through graphitic carbon oxidative dissolution (see Tumiati et al., 2020). The welded-shut Au60Pd40 capsule was enclosed in a larger Au capsule (outer diameter: 4.5 mm; inner diameter: 4.3 mm; length: 9–10 mm) containing a fayalite–magnetite–quartz–water (FMQ) fO2/fH2 buffer in order to control the redox potential during the experimental run. Such relatively oxidizing conditions were necessary to obtain CO2 as the dominant volatile carbon species in the COH fluid (e.g., Connolly, 1995). Note that at our experimental pressure and temperature conditions the assemblage fayalite + magnetite + quartz reacts to form ferrosilite + magnetite + coesite. This imposes fO2 that is 0.8 log units higher compared to the metastable assemblage fayalite + magnetite + quartz (ΔFMQ ). Details of buffer preparation, buffering process, and relevant reactions can be found in Toffolo et al. (2023) and Tumiati et al. (2020). The weld-sealed double capsule was placed in the center of a 34 mm long assembly composed of an inner MgO rod (diameter: 6 mm), a graphite furnace (outer diameter: 8 mm), and an outer NaCl sleeve (pressure medium; outer diameter: 12.6 mm). The run temperature was monitored using a sheathed (outer steel sheath plus alumina ceramic insulator) K-type thermocouple (with a precision of ±5 °C) inserted in the MgO rod and separated from the capsule by a 0.6 mm thick polycrystalline Al2O3 disk. The experiments were run using a 600 tonne rocking end-loaded piston–cylinder apparatus (Experimental Petrology Laboratory, DiSTAD, University of Milan). A pressure calibration relying on the quartz-to-coesite phase transition curve (Bose and Ganguly, 1995) enables an accuracy of ±0.1 GPa. The experiments lasted 168 h (7 d) and were terminated by switching off the power supply, ensuring a temperature drop at a rate >40 °C s−1. In the case of C–O–H fluids, this rate is sufficiently high to preserve the high-pressure, high-temperature fluid speciation (Tiraboschi et al., 2016). At the end of the experiment the double capsule was cleaned in a bath of HCl–H2O 1:20 solution to remove any residue of MgO, and then the outer Au capsule was partly opened to expose the inner Au60Pd40 capsule and heated in a vacuum oven at 110 °C for 30 min to remove the residual water impregnating the buffer assemblage. The dried capsule was partly embedded in epoxy resin, leaving the opened part exposed to the surface to enable the piercing and release of volatiles for subsequent analysis.
2.2 Sampling and analysis of the COH volatile phase
In order to extract and analyze the COH volatile phase the experimental capsules were mounted in a capsule-piercing device connected to a quadrupole mass spectrometer (Experimental Petrology Laboratory, DiSTAD, University of Milan). After the measurement, the run products – specifically, the volatile phase alone in OAA decomposition experiments, as well as both the volatile phase and solid residue in organic matter pyrolysis experiments – were recovered to determine their isotopic signature by means of an isotope ratio mass spectrometer (Stable Isotopes Laboratory, DiSTAD, University of Milan).
2.2.1 Volatile-phase extraction
The capsule-piercing device, developed by Tiraboschi et al. (2016), which consists of a PTFE reactor embedded in an electric furnace (Fig. 1), was used to sample the COH volatile phase from the experimental capsules. The epoxy mount containing the capsule is placed in a sample holder that is integrated with the hollow base of the reactor. Using a wrench to turn the base (with each full rotation corresponding to a 2 mm vertical movement), the capsule is gradually brought closer to a fixed steel end-mill cutter, which finally pierces it. Upon puncturing, the gas contained in the capsule fills the reactor, which is maintained at a temperature of ∼ 90 °C, necessary to let water completely evaporate. On the top of the reactor two three-way valves (V3 and V4 in Fig. 1) control the inlet for Ar carrier gas and the outlet of the gas mixture to be analyzed by the mass spectrometer. The gas flows from the reactor through a stainless steel piping to a three-way needle valve (V7), where a small flux is sampled by the spectrometer line; the largest part of the gas is conveyed to another three-way valve (V8) connecting the main line to either a vacuum pump or the vent line. The vacuum pump is only used in preliminary cleaning of the line and the reactor; in successive operations it is excluded from the line (Fig. 1a). The line segment that goes from the reactor to the spectrometer is heated to 105 °C by a winded resistance to prevent water condensation. The carrier gas flux is regulated by a Bronkhorst® mass flow controller. The pressure in the line and in the reactor is measured by two Baumer® high-sensitivity transducers having an error of ±1 mbar. The temperatures of the line, the reactor, and the furnace are provided by three K-type thermocouples. An Eurotherm® PID recorder and controller allows the monitoring of the temperatures and pressures and automatically performs temperature adjustments. When OAA decomposition is conducted in an inline tube oven (see experiment OAA_5), the reactor is bypassed, and the volatile phase is sampled by simultaneously opening valves V5 and V6 immediately after the experimental run ends.
2.2.2 Quadrupole mass spectrometry
The composition of the COH volatile phase was determined using an Extorr Inc. XT200 quadrupole mass spectrometer (QMS) equipped with a secondary electron multiplier. The spectrometer operates at high vacuum ( mbar), which is obtained through a turbomolecular pump coupled with a backing pump (Edwards® T-Station 85). The QMS simultaneously measures 13 channels through a Faraday cup, covering the fragmentation products of key volatile species (CH4, CO, CO2, H2, H2O, N2, O2), with signal collection lasting 1554 s. Signal intensity is enhanced by inserting a secondary electron multiplier during both background and sample acquisitions. Data are presented as time (s) versus signal intensity (arbitrary unit) elution profiles. The integration and the conversion of the profiles to micromoles is done using a dedicated Wolfram Mathematica® routine incorporating a calibration matrix. To address instrumental drift, the calibration is periodically updated using measurements of pure water, air, and specific gas mixtures: (1) Ar + CO2 (10 vol. %) + O2 (9.85 vol. %); (2) Ar + H2 (10 vol. %); (3) Ar + CH4 (10 vol. %) + CO (10 vol. %). Measurement accuracy is ensured by analyzing the volatiles (H2O, CO, CO2, and H2) generated by the thermal decomposition at 250 °C of 1 mg of oxalic acid dihydrate (Tiraboschi et al., 2016). Relative uncertainties in the measurements are estimated at approximately 1 % for H2O, CO2, CH4, H2, and O2, except for CO, which exhibits an uncertainty of ∼ 10 % due to interference with N2 from the air on the channel .
2.2.3 CO2 sampling
During QMS analysis, only a small fraction of the carrier gas and sample mixture is directed to the spectrometer through a needle valve V7 and a quartz capillary tube, while the remainder is released into the atmosphere through the vent line (Fig. 1a). Consequently, we developed a procedure that involves the sampling of the otherwise discarded gas. The gas flow is redirected at the three-way valve V8 from the vent to the line of the vacuum pump, which has been previously disconnected (Fig. 1b). A syringe needle with a shaft length of 95 mm and a lumen diameter of 0.85 mm is attached to the end of the tube. To sample the gas, the needle is inserted through a silicone septum into the bottom of an Exetainer® 12 mL borosilicate glass test tube, previously filled with pure He at 1 bar (Fig. 1c). Just before inserting the needle, the septum is punctured by another smaller syringe needle (15 mm long, with a lumen diameter of 0.45 mm) to prevent pressure buildup in the test tube. The 12 mL test tube is filled within 70 s at the specified carrier gas flow rate of 10 mL min−1. Upon completion of the sampling, the longer needle is extracted before removing the shorter one. The need to avoid overpressurization arises from the requirement to introduce minimal amounts of gas into the IRMS to ensure optimal sensitivity. To maximize the quantity of sampled CO2, the volume of gas that produced the peak in the QMS elution profile must be intercepted.
2.2.4 Isotope ratio mass spectrometry
The δ13C signature (standardized to VPDB) of CO2 was determined using a Thermo Scientific™ Delta V Advantage isotope ratio mass spectrometer (IRMS) equipped with a GasBench II gas preparation and introduction device operating in continuous flow mode. In the GasBench II, an autosampler collects the CO2-bearing gaseous sample contained in a gas-tight vial using a He carrier gas flux. The gas mixture first flows through a water diffusion trap composed of a glass tube containing a Nafion™ membrane, and then it reaches a 100 µL loop injection system, where an eight-port Valco® valve loads and injects the gas into a PoraPLOT Q™ isothermal gas chromatography (GC) column (capillary length: 25 m; inner diameter: 0.32 mm; film thickness: 10 µm) in order to separate different molecular species. The effluent gas, before entering the spectrometer, is conveyed through a second water trap to an active open-split interface, where it can be automatically diluted with He if the first signal pulse exceeds a threshold of 10 000 mV. In the spectrometer, the gas is ionized through an electron impact source. The spectrometer takes advantage of five Faraday cup detectors: two are dedicated to measuring the masses at 2 (H) and 3 (HD+); a universal triple collector collects the signal associated with the masses at 44 (12C16O, 14NO+), 45 (13C16O, 12C16O17O+, 15N14N16O+, 14NO+), and 46 (12C16O18O+, 12C17O, 14NO+, 15NO+, 14N15N17O+). The isotopic characterization of the sample is achieved by referencing it to a pure (99.998 %) CO2 working gas, which is separately introduced into the spectrometer through an injection system, featuring an active open-split interface. A typical continuous-flow IRMS chromatogram comprises 13 consecutive signal pulses: the first 4 relate to the working gas, while the remaining 9 pertain to the sample. The signal is reported as voltage, and values below 1000 mV cannot be reliably quantified using our analytical setup. Signal acquisition and processing, including the conversion to δ13C signatures, are obtained using the Isodat software suite. The accuracy of the IRMS is checked periodically by analyzing the δ13C of CO2 produced by the digestion with phosphoric acid of calcium carbonate in-house standards (Merck® carbonate: δ13C ; MAQ Carrara marble: δ13C ).
The δ13C of bulk OAA and Tetraselmis suecica, as well as of the residual char contained in the capsules of experiments COH210 and COH245, was determined by connecting the IRMS with a Thermo Scientific™ Flash 2000 organic elemental analyzer (OEA) set to nitrogen and carbon (NC) mode. In this configuration, the sample contained in a Sn foil capsule is inserted by an autosampler in a combustion reactor (a quartz tube containing CuO, as oxidizing agent, and silvered Co(II,III) oxide, as sulfur trap) at 950 °C. A 10 s flux of oxygen (250 mL min−1) fully oxidizes the sample. The resultant gases are conveyed by He carrier gas to a second reactor (a quartz tube containing metallic copper) at 840 °C where reduction of nitrogen oxides to N2 and filtering of excess O2 occur. The gas mixture then flows through a glass absorption filter containing magnesium perchlorate to trap the residual H2O and is injected into a GC column (2 m stainless steel tube) to separate CO2 from N2. A thermal conductivity detector (TCD) measures the total carbon content of the sample, after which CO2 is directed through a Thermo Scientific™ ConFlo IV interface to the IRMS for isotopic characterization. OEA peripheral control and data-handling are performed with EagerSmart™ software.
2.3 Preservation of the redox buffer assemblage
In the double-capsule experiments involving the pyrolysis of microalgae, the buffering of the redox state is effective as long as the entire assemblage (ferrosilite + magnetite + quartz + water) is preserved throughout the run. The preservation of the buffer is assessed in two steps. First, the presence of water in the buffer is evaluated upon opening the outer gold capsule. As a qualitative indicator, water typically gushes out when the capsule is pricked. If this does not occur, the capsule is weighed immediately after opening and again after drying in a vacuum oven to detect any weight loss attributable to water. Second, after the capsule is pierced and the volatile analysis is conducted, the solid assemblage is examined. Standard 25.4 mm (1 in) epoxy mounts are prepared and polished to expose the redox buffer assemblage. Mineral composition is determined using a JEOL 8200 wavelength-dispersive electron microprobe (DiSTAD, University of Milan), operating at 15 kV accelerating potential, 5 nA sample current, and 1 µm beam size. Fayalite is used as the standard for Fe and grossular for Si, with a counting time of 30 s (including 10 s for the background) for all elements.
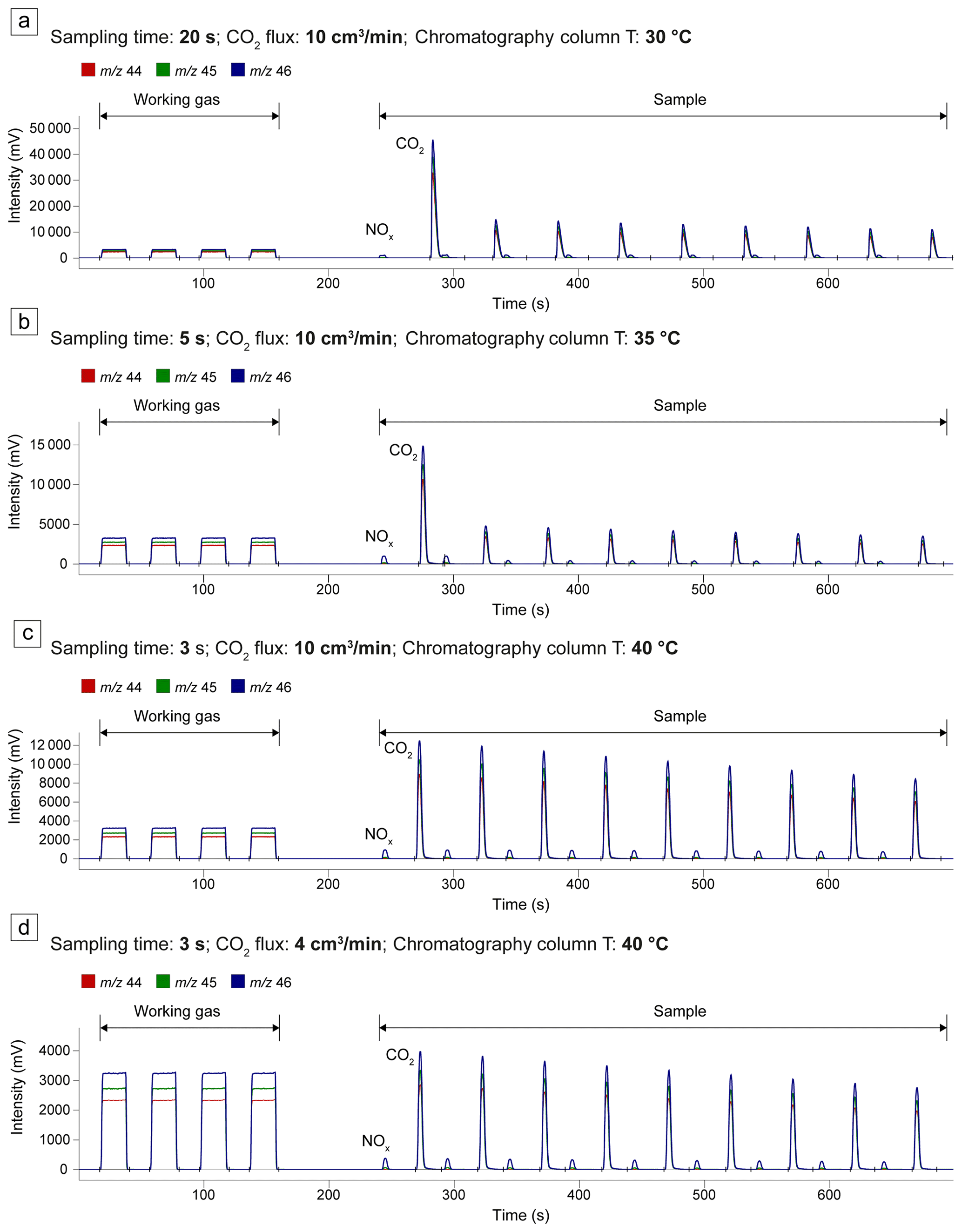
Figure 3IRMS chromatograms obtained by varying the flux of the QMS CO2 calibration mixture in the line, the sampling time, and the temperature of the chromatography column. See the main text for a detailed explanation.
3.1 Preparatory IRMS tuning and fractionation test
The effective separation of the contributions of CO2 and NOx to the channels 44, 45, and 46 is essential in every IRMS analysis. Nitrogen, in fact, is a ubiquitous contaminant in the analyzed gases: it is present during capsule preparation and CO2 sampling, both being performed in air, and it is contained as trace amounts in the Ar carrier gas of the QMS line. Additionally, the accuracy of the analysis conducted by the IRMS improves when the signal intensity of the sample closely matches that of the working gas. To determine the optimal conditions, four tests were conducted in which we analyzed the CO2 calibration mixture of the QMS. To replicate the gas sampling procedure employed for the experimental capsules, the calibration gas flowed through the QMS line and was then sampled according to the procedure outlined in Sect. 2.2.3. In these tests, the following parameters were adjusted: the flux of the calibration gas in the QMS line, the duration of sampling from the line into the glass test tube, and the temperature of the IRMS GC column. The results are reported in Fig. 3. In each sample analysis the more intense signal pulses correspond to CO2, while the less intense relate to nitrogen oxides (NOx). In the first two tests, the temperature of the chromatography column was too low (30 and 35 °C, respectively) to achieve adequate separation between NOx and CO2 (Fig. 3a, b). Moreover, in both cases, the signal intensity was excessively high (≫10 000 mV) due to prolonged sampling from the QMS line (20 and 5 s, respectively), prompting the automatic dilution system of the IRMS (see Sect. 2.2.4). Consequently, in the third test, the temperature of the column was raised to 40 °C, and the sampling duration was further reduced to 3 s (Fig. 3c). While satisfactory peak separation was attained, the signal (>10 000 mV) remained too high compared to the working gas (∼ 3000 mV). Hence, in the fourth test, the flux of the QMS calibration mixture was decreased from 10 to 4 cm3 min−1, while the column temperature and sampling duration remained the same as in the third test. This final test yielded optimal conditions of signal intensities (∼ 3000 mV) and peak separation (Fig. 3d). Using these conditions and the CO2 sampling procedure described in Sect. 2.2.3, we measured the δ13C of the QMS CO2 calibration mixture directly from the cylinder (Table S1), maintaining the QMS line at room temperature (28 °C). The δ13C was . To test for potential temperature-induced fractionation effects in the QMS line, we repeated the measurements with the line temperature set to 105 °C, the standard temperature for QMS analysis. This resulted in a very similar δ13C of .
Table 2Composition of the volatile phase and carbon isotopic signature of CO2 from OAA decomposition experiments. The bulk δ13C of OAA is . The numbers in brackets refer to the analytical errors, expressed as 1σ on the last digits, with the exception of CO2/CO and XCO2, where the error is obtained by propagation of the uncertainties associated with the individual species.
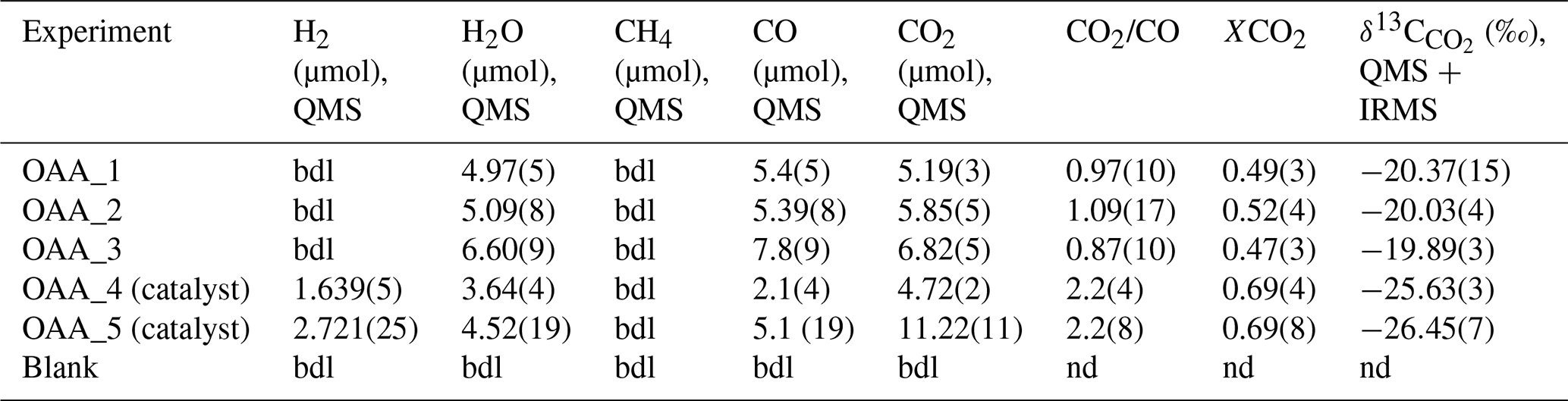
bdl: below detection limit; nd: not determinable.
3.2 Chemical and isotopic characterization of experimental CO2-bearing COH fluids
3.2.1 Anhydrous oxalic acid decomposition experiments
All the product COH fluids consist of a mixture of H2O, CO2, and CO, but the proportions of the volatile species, the H2 content, and the δ13C appear to be dependent on the presence of a catalyst (Table 2 and Tables S2 and S3 in the Supplement).
Experiments without catalyst
In the three experiments performed without the Pt catalyst, the proportion among H2O, CO2, and CO is approximately and no H2 has been detected. The ratios between CO2 and CO and the CO2 molar fraction, XCO2 [= CO2/(CO2+ CO)]molar, are in the range ∼ 0.9–1.1 and ∼ 0.47–0.52, respectively. The δ13C is very similar within uncertainty and averages at .
Experiments with catalyst
In the experiment OAA_4 and in control experiment OAA_5, where a Pt catalyst was used, up to 3 µmol of H2 is produced; moreover, the CO2 CO ratio and XCO2 are ∼ 2.2 and 0.69 in both experiments. However, in experiment OAA_4 an excess amount of water is observed compared to the composition expected from the decomposition of OAA (Fig. S1). Nonetheless, δ13C values in the two experiments are comparable and average at .
In the blank experiment the QMS could detect only N2 and O2 (not reported), the main components of air; also, the IRMS could not determine the δ13C of the air enclosed in the capsule due to the extremely low signal on the three channels ( 44 =12 mV; 45 =15 mV; 46 =20 mV).
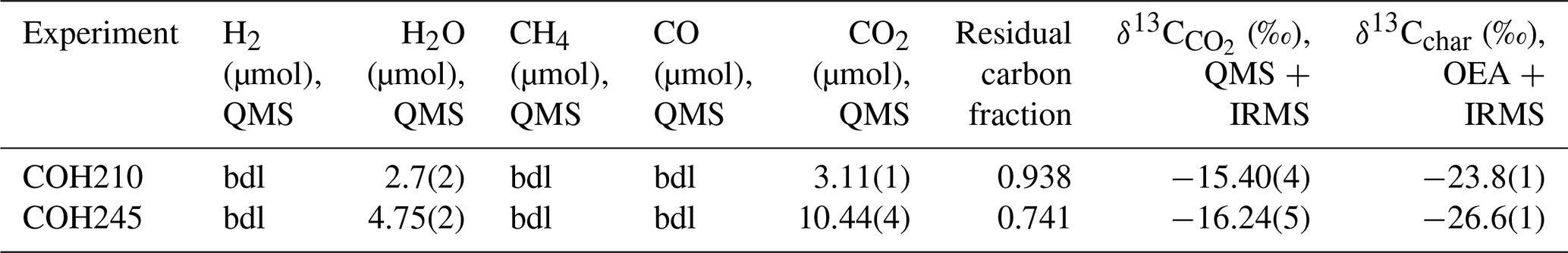
Table 3Composition of the volatile phase, residual carbon fraction, and isotopic signature of both CO2 and char produced from green algae (Tetraselmis suecica) pyrolysis experiments at 3 GPa, 700 °C, and ΔFMQ . The bulk δ13C of green algae is . The numbers in brackets refer to the analytical errors, expressed as 1σ on the last digit.
3.2.2 Redox-buffered, high-pressure, high-temperature organic matter pyrolysis experiments
In the two experiments carried out at 3 GPa, 700 °C, and ΔFMQ (COH210, COH245), a COH fluid consisting of CO2 and H2O was generated (Tables 3, S4, S5). The experiment COH245 contains more water than COH210 (10 µmol against 3 µmol, respectively), since in the former water was added as a starting material.
The measured δ13C values are and for COH210 and COH245, respectively.
4.1 Reproducibility and accuracy
The QMS analysis of the OAA decomposition products suggests that no mass loss or alteration in volatile proportion occurred during the experiment or through the sampling process (Fig. S1). The experiment duration of 1 h prevented significant H2 loss from the Au capsule, an issue observed by Tiraboschi et al. (2016) during longer experiments using oxalic acid dihydrate. Only one experiment (OAA_4) shows an excess of ∼ 1.5 µmol of H2O compared to the expected volatile proportion from the decomposition of stoichiometric OAA. This anomaly is likely due to H2O absorbed by the reagent rather than accidental infiltration of humidity during sampling. Nevertheless, this minor water contamination does not affect subsequent carbon isotopic determinations. The systematic difference in δ13C values between the two sets of OAA decomposition experiments – with and without a catalyst ( and , respectively) – is noteworthy. Isotopic fractionation or contamination during the sampling of the volatile phase can be excluded for the following reasons. First, in the test where the CO2 calibration mixture was directly sampled from the cylinder, with the QMS line maintained at two different temperatures (28 and 105 °C; Sect. 3.1), two very similar δ13C values for CO2 were obtained (δ13C and δ13C ), indicating that temperature-induced carbon isotopic fractionation during sampling of the volatile phase is negligible. Second, in the blank experiment, containing only air in the capsule, volatile carbon species were undetectable by the QMS (≪1 µmol), and the low signal associated with CO2 isotopologues (≤20 mV) prevented a δ13C determination by the IRMS, ruling out any significant contribution to δ13C from CO2 in the air, either trapped in experimental capsules or infiltrated into the QMS line during sampling. Additionally, periodic analysis of calcium carbonate standards further ensures no drift in the IRMS results. Remarkably, catalyst-assisted decomposition produced CO2 with a δ13C value identical to the bulk δ13C of OAA (), whereas experiments without the catalyst yielded higher δ13C, indicating, in the latter case, a stronger fractionation effect between coexisting CO2 and CO. The fluids produced in the two sets of experiments also differed, with the catalyst-assisted experiments being enriched in H2 and showing higher and less scattered CO2 / CO (∼ 2.2) and XCO2 (∼ 0.69). As noted by Sackett and Chung (1979), at such low temperatures and short experiment durations, isotopic exchange between volatile carbon species is inhibited, and the isotopic abundances are governed by reaction kinetics (i.e., kinetic isotope effect, KIE; see also Kueter et al., 2020, and references therein). According to Sato et al. (2002), at low temperatures (<650 °C) the conversion of CO to CO2 via the water–gas shift reaction (CO + H2O = CO2+ H2) results in isotopically heavier residual CO, yielding an apparent fractionation factor approaching 0 at 300 °C. In agreement with this, fractionation factors between coexisting CO2 and CO () calculated for our catalyst-assisted experiments are close to 0 (Table 4; see Sect. 4.2.1). Therefore, the KIE associated with a different progression of the water–gas shift reaction may, at least in part, explain our results. A further investigation of the causes of carbon fractionation, however, is beyond the scope of this study. Nonetheless, it is important to note that both groups of experiments produced consistently similar δ13C ratios with low standard deviations (σ=0.6 ‰ and 0.2 ‰ for the sets of experiments with and without a catalyst, respectively). Therefore, rounding up the uncertainty, the precision of our method can be conservatively estimated at around 1 ‰.
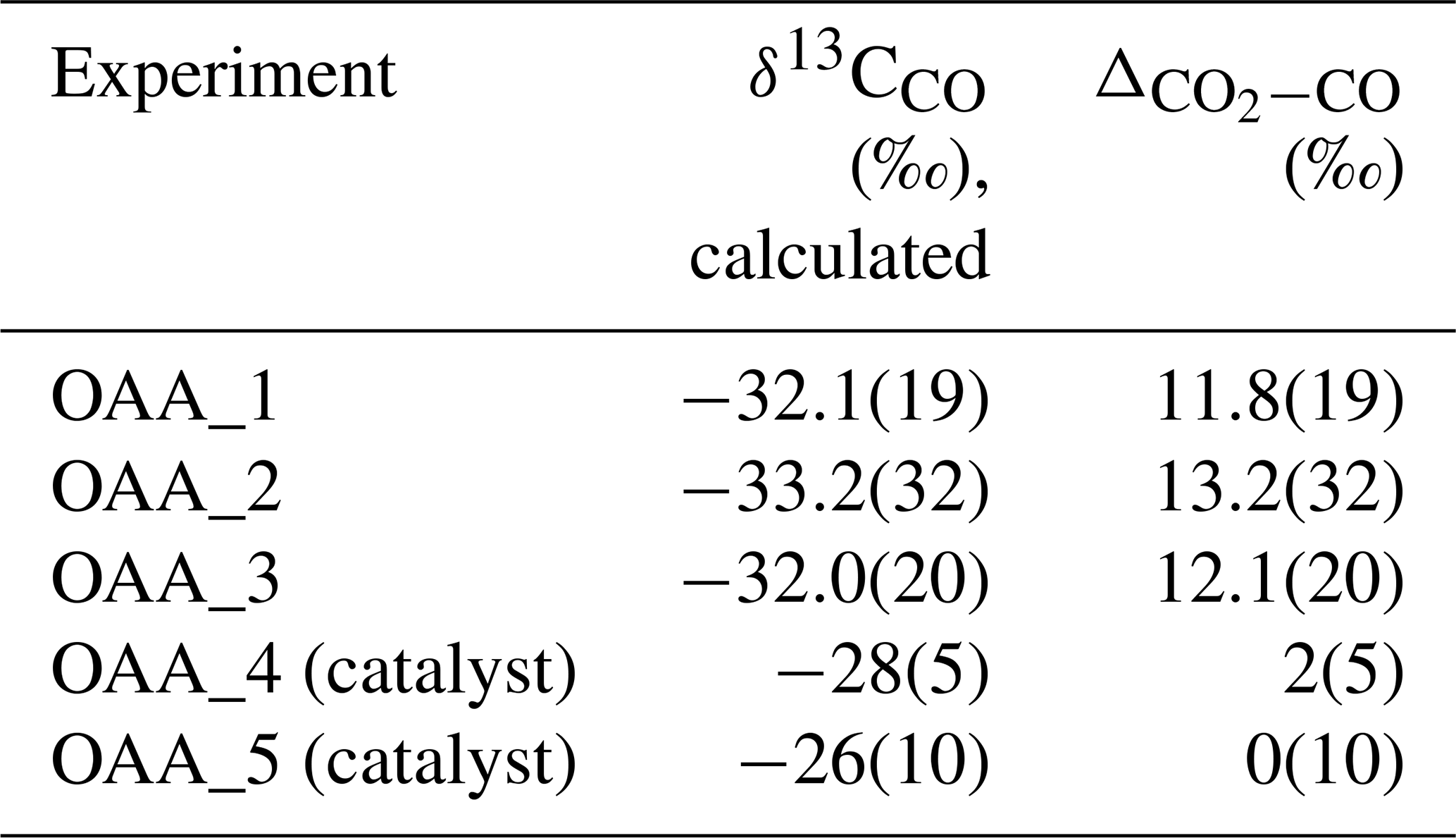
Table 4Calculated δ13C of CO and CO2–CO fractionation factors for OAA decomposition experiments. The numbers in brackets indicate the 1σ uncertainty on the last digits.
4.2 Potential applications
4.2.1 Fractionation factor calculation
Our method allows for the determination of both the composition of the volatile phase and the δ13C from the same sample contained in experimental capsules. These data can be combined in the following mass balance equation:
where the δ13Cbulk of OAA is known () to determine the δ13C for CO and, consequently, the (Table 3). In our OAA decomposition experiments, we obtain an average δ13CCO of and a of 12.4±1.4 ‰ for catalyst-free experiments, while average δ13CCO and are and 1±5 ‰, respectively, for catalyst-assisted experiments. These values differ significantly from the equilibrium reported by Kueter et al. (2019a) at 250 °C, i.e., 34±1 ‰. However, given the uncertainty regarding equilibrium attainment at 250 °C after only 1 h and the effect of the catalyst on both reaction progress and isotopic exchange, our fractionation factor should be interpreted as an apparent value. Nevertheless, with specifically designed experiments, it should be possible to determine equilibrium fractionation factors between coexisting volatile carbon pairs (e.g., CO2–CO, CO2–CH4) under geologically relevant conditions. Moreover, providing the IRMS with specific working gases would allow for the direct δ13C determination of the other volatile carbon species.
4.2.2 Assessment of equilibrium attainment in petrological experiments involving a CO2-bearing volatile phase
In both double-capsule experiments performed at 3 GPa, 700 °C, and ΔFMQ (COH210, COH245), we were able to determine the volatile composition of the fluid as well as the δ13C of CO2. Although the fluids in both experiments were compositionally similar, i.e., CO2+ H2O mixtures, the CO2 H2O ratios were approximately 1 in COH210 and 2 in COH245. This difference likely arises because in COH210, both water and CO2 originated solely from the pyrolysis of organic molecules, whereas in COH245 water was added as a starting material, prompting a more extensive oxidative dissolution of organic matter into CO2. Consequently, the percentage of carbon converted to CO2 was 6.2 % in COH210 and 25.9 % in COH245. The residual char also exhibited distinct δ13C values in the two experiments: δ13C in COH210 and δ13C in COH245. The decrease in both δ13C and δ13Cchar along with the remaining fraction of carbon is consistent with a model of closed-system isotopic fractionation from graphite to CO2 (Fig. S2). This model was calculated using the software “Thermotopes” (Boutier et al., 2024), selecting a fractionation factor of 7.7 ‰, as determined by Bottinga (1969). For graphite with an initial δ13C of −23.4 ‰ (corresponding to δ13Cbulk of Tetraselmis suecica) and with residual carbon fractions of 0.938 or 0.741, the calculated δ13C ratios are δ13C and δ13C or δ13C and δ13C, respectively. The good correspondence between our results and the model has two key implications. First, our method of CO2 sampling and analysis is a reliable tool for fully characterizing the volatile composition and the natural-like δ13C of CO2-bearing volatile phases, enabling us to potentially assess equilibrium attainment during redox-buffered, high-pressure, and high-temperature petrological experiments. Second, using organic matter as a source of carbon, rather than graphite itself, proved to be an effective approach for producing a CO2-bearing volatile phase that is close to isotopic equilibrium with solid carbon. Previous studies have shown that at up to ∼ 700 °C graphite has sluggish reactivity and tends to dissolve irreversibly (Toffolo et al., 2023; Tumiati et al., 2022; Ziegenbein and Johannes, 1980). This, combined with very low carbon diffusivity, results in isotopically zoned graphite where inner portions retain the initial δ13C and only the rims participate in carbon isotope exchange when CO2 or other volatile carbon species are produced (Kueter et al., 2019b; Scheele and Hoefs, 1992; Tumiati et al., 2022). However, our experiments suggest enhanced isotope exchange between char and CO2. A possible explanation is that since both char and CO2 formed contemporaneously from organic matter, isotope exchange between the two phases could occur. To our knowledge, no experimental data currently exist on this process; therefore a dedicated study is needed for a deeper understanding.
In this paper we demonstrated the possibility of integrating an IRMS with the capsule-piercing device to analyze the volume of gas that is discarded during standard QMS analysis of a volatile phase produced in petrological experiments. This setup enables the simultaneous determination of both the volatile composition and carbon isotopic signature of CO2-bearing COH fluids. The measured δ13C ratios exhibit high reproducibility and accuracy (within 1 ‰) and are not significantly affected by fractionation processes during sampling. The data obtained through this method can be used to calculate the δ13C of volatile species coexisting with CO2 and the fractionation factors governing the isotopic exchange. Furthermore, this approach can be combined with standard analyses of solid carbon – in the form of graphite/diamond but also carbonate – to provide a comprehensive scenario of carbon isotope exchange, allowing for evaluating the attainment of chemical and isotopic equilibrium between solid carbon and COH fluids. Ultimately, this method opens the possibility of designing experiments dedicated to exploring the partitioning of carbon isotopes among various reservoirs within the Earth's interior.
The Wolfram Mathematica® code used to process raw QMS data is available upon request from the authors.
All the data related to this article can be found in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/ejm-37-25-2025-supplement.
LT: writing (original draft preparation), formal analysis, investigation, conceptualization. LM: investigation, writing (review and editing). EF: investigation, formal analysis, writing (review and editing). ST: methodology, conceptualization, writing (review and editing), investigation, software, funding acquisition.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This article is part of the special issue “Probing the Earth: experiments on and for our planet”. It is a result of the EMPG 2023 conference, Milan, Italy, 12–15 June 2023.
Sandro Recchia (DiSAT, University of Insubria) is sincerely acknowledged for his precious technical support in setting up the gas sampling and analytical apparatus. We thank Chiara Compostella (DiSTAD, University of Milan) for the OEA analyses. We are grateful to Francois Holtz, Nico Küter, and an anonymous reviewer for their constructive feedback and valuable suggestions.
This research has been supported by the Italian program MIUR PRIN (project HYDECARB, grant ID 20224YR3AZ) and MIUR project “Dipartimenti di Eccellenza 2023–2027”. Luca Toffolo has been supported by the Special Young Researcher Promotion 2021–2024 by the SIMP (Società Italiana di Mineralogia e Petrologia) and the APC central fund of the University of Milan to cover publication costs.
This paper was edited by Francois Holtz and reviewed by Nico Küter and one anonymous referee.
Bose, K. and Ganguly, J.: Quartz-coesite transition revisited: reversed experimental determination at 500–1200 ° C and retrieved thermochemical properties, Am. Mineral., 80, 231–238, https://doi.org/10.2138/am-1995-3-404, 1995.
Bottinga, Y.: Calculated fractionation factors for carbon and hydrogen isotope exchange in system calcite–carbon dioxide–graphite–methane–hydrogen–water vapor, Geochim. Cosmochim. Ac., 33, 49–64, https://doi.org/10.1016/0016-7037(69)90092-1, 1969.
Boutier, A., Martinez, I., Daniel, I., Tumiati, S., Siron, G., and Brovarone, A. V.: Thermotopes-COH–A software for carbon isotope modeling and speciation of COH fluids, Comput. Geosci., 184, 105533, https://doi.org/10.1016/j.cageo.2024.105533, 2024.
Brand, W. A., Douthitt, C. B., Fourel, F., Maia, R., Rodrigues, C., Maguas, C., and Prohaska, T.: Gas Source Isotope Ratio Mass Spectrometry (IRMS), in: Sector Field Mass Spectrometry for Elemental and Isotopic Analysis, edited by: Prohaska, T., Irrgeher, J., Zitek, A., and Jakubowski, N., Roy. Soc. Ch., https://doi.org/10.1039/9781849735407-00500, 500–549, 2015.
Connolly, J. A. D.: Phase diagram methods for graphitic rocks and application to the system C−O−H−FeO−TiO2−SiO2, Contrib. Mineral. Petr., 119, 94–116, https://doi.org/10.1007/BF00310720, 1995.
Eggler, D. H., Mysen, B. O., and Hoering, T. C.: The solubility of carbon monoxide in silicate melts at high pressures and its effect on silicate phase relations, Earth Planet. Sc. Lett., 43, 321–30, https://doi.org/10.1016/0012-821X(79)90218-8, 1979.
Eugster, H. P.: Heterogeneous reactions involving oxidation and reduction at high pressures and temperatures, J. Chem. Phys., 26, 1760–1761, https://doi.org/10.1063/1.1743626, 1957.
Eugster, H. P. and Skippen, G. B.: Igneous and metamorphic reactions involving gas equilibria, Res. Geochem., 2, 492–520, 1967.
Horita, J.: Carbon isotope exchange in the system CO2-CH4 at elevated temperatures, Geochim. Cosmochim. Ac., 65, 1907–1919, https://doi.org/10.1016/S0016-7037(01)00570-1, 2001.
Kueter, N., Schmidt, M. W., Lilley, M. D., and Bernasconi, S. M.: Experimental determination of equilibrium CH4–CO2–CO carbon isotope fractionation factors (300–1200 ° C), Earth Planet. Sc. Lett., 506, 64–75, https://doi.org/10.1016/j.epsl.2018.10.021, 2019a.
Kueter, N., Lilley, M. D., Schmidt, M. W., and Bernasconi, S. M.: Experimental carbonatite/graphite carbon isotope fractionation and carbonate/graphite geothermometry, Geochim. Cosmochim. Ac., 253, 290–306, https://doi.org/10.1016/j.gca.2019.03.020, 2019b.
Kueter, N., Schmidt, M. W., Lilley, M. D., and Bernasconi, S. M.: Kinetic carbon isotope fractionation links graphite and diamond precipitation to reduced fluid sources, Earth Planet. Sc. Lett., 529, 115848, https://doi.org/10.1016/j.epsl.2019.115848, 2020.
Luque, F. J., Crespo-Feo, E., Barrenechea, J. F., and Ortega, L.: Carbon isotopes of graphite: implications on fluid history, Geosci. Front., 3, 197–207, https://doi.org/10.1016/j.gsf.2011.11.006, 2012.
Mason, E., Edmonds, M., and Turchyn, A. V.: Remobilization of crustal carbon may dominate volcanic arc emissions, Science, 357, 290–294, https://doi.org/10.1126/science.aan5049, 2017.
Meier-Augenstein, W.: Applied gas chromatography coupled to isotope ratio mass spectrometry, J. Chromatogr. A, 842, 351–371, https://doi.org/10.1016/S0021-9673(98)01057-7, 1999.
Miozzi, F. and Tumiati, S.: Aqueous concentration of CO2 in carbon-saturated fluids as a highly sensitive oxybarometer, Geochem. Perspec. Lett., 16, 30–34, https://doi.org/10.7185/GEOCHEMLET.2040, 2020.
Peng, W., Tumiati, S., Zhang, L., Tiraboschi, C., Vitale Brovarone, A., Toffolo, L., and Poli, S.: An experimental study on kinetics-controlled Ca-carbonate aqueous reduction into CH4 (1 and 2 GPa, 550 ° C): Implications for C mobility in subduction zones, J. Petrol., 63, egac070, https://doi.org/10.1093/petrology/egac070, 2022.
Rosenbaum, J. M. and Slagel, M. M.: COH speciation in piston-cylinder experiments, Am. Mineral., 80, 109–14, https://doi.org/10.2138/am-1995-1-211, 1995.
Sackett, W. M. and Chung, H. M.: Experimental confirmation of the lack of carbon isotope exchange between methane and carbon oxides at high temperatures, Geochim. Cosmochim. Ac., 43, 273–276, https://doi.org/10.1016/0016-7037(79)90246-1, 1979.
Sato, M., Mori, T., Shimoike, Y., Nagao, K., and Notsu, K.: Carbon isotope systematics of CO2, CO and CH4 in fumarolic gases from Satsuma-Iwojima volcanic island, Japan, Earth Planets Space, 54, 257–263, https://doi.org/10.1186/BF03353025, 2002.
Scheele, N. and Hoefs, J.: Carbon isotope fractionation between calcite, graphite and CO2: an experimental study, Contrib. Mineral. Petr., 112, 35–45, https://doi.org/10.1007/BF00310954,1992.
Stachel, T., Chacko, T., and Luth, R. W.: Carbon isotope fractionation during diamond growth in depleted peridotite: Counterintuitive insights from modelling water-maximum CHO fluids as multi-component systems, Earth Planet. Sc. Lett., 473, 44–51, https://doi.org/10.1016/j.epsl.2017.05.037, 2017.
Tiraboschi, C., Tumiati, S., Recchia, S., Miozzi, F., and Poli, S.: Quantitative analysis of COH fluids synthesized at HP–HT conditions: An optimized methodology to measure volatiles in experimental capsules, Geofluids, 16, 841–855, https://doi.org/10.1111/gfl.12191, 2016.
Tiraboschi, C., Tumiati, S., Sverjensky, D., Pettke, T., Ulmer, P., and Poli, S.: Experimental determination of magnesia and silica solubilities in graphite-saturated and redox-buffered high-pressure COH fluids in equilibrium with forsterite+ enstatite and magnesite+ enstatite, Contrib. Mineral. Petr., 173, 1–17, https://doi.org/10.1007/s00410-017-1427-0, 2018.
Tiraboschi, C., Miozzi, F., and Tumiati, S.: Carbon-saturated COH fluids in the upper mantle: a review of high-pressure and high-temperature ex situ experiments, Eur. J. Mineral., 34, 59–75, https://doi.org/10.5194/ejm-34-59-2022, 2022.
Toffolo, L., Tumiati, S., Villa, A., Fumagalli, P., Amalfa, A., and Miozzi, F.: Experimental dissolution of carbonaceous materials in water at 1 GPa and 550° C: Assessing the role of carbon forms and redox state on COH fluid production and composition during forearc subduction of organic matter, Front. Earth Sci., 11, 1013014, https://doi.org/10.3389/feart.2023.1013014, 2023.
Tumiati, S., Tiraboschi, C., Sverjensky, D. A., Pettke, T., Recchia, S., Ulmer, P., Miozzi, F., and Poli S.: Silicate dissolution boosts the CO2 concentrations in subduction fluids, Nat. Commun., 8, 616, https://doi.org/10.1038/s41467-017-00562-z, 2017.
Tumiati, S., Tiraboschi, C., Miozzi, F., Vitale-Brovarone, A., Manning, C. E., Sverjensky, D. A., Milani, S., and Poli, S.: Dissolution susceptibility of glass-like carbon versus crystalline graphite in high-pressure aqueous fluids and implications for the behavior of organic matter in subduction zones, Geochim. Cosmochim. Ac., 273, 383–402, https://doi.org/10.1016/j.gca.2020.01.030, 2020.
Tumiati, S., Recchia, S., Remusat, L., Tiraboschi, C., Sverjensky, D. A., Manning, C. E., Vitale Brovarone, A., Boutier, A., and Spanu, D. and Poli, S.: Subducted organic matter buffered by marine carbonate rules the carbon isotopic signature of arc emissions, Nat. Commun., 13, 2909, https://doi.org/10.1038/s41467-022-30421-5, 2022.
Ziegenbein, D. and Johannes, W.: Graphite in C-H-O fluids: An unsuitable compound to buffer fluid composition at temperatures up to 700 ° C, Neues Jb. Miner. Monat., 7, 289–305, 1980.
This study introduces an innovative method to analyze carbon isotopes in tiny amounts of CO2 produced during experiments that simulate conditions deep within the Earth. Using advanced tools, we measured both the composition and isotopic signature of CO2 in carbon-bearing fluids. This enables new studies of carbon exchange between rocks and fluids under extreme conditions, offering new insights into processes driving the deep carbon cycle and, ultimately, controlling the climate of our planet.
This study introduces an innovative method to analyze carbon isotopes in tiny amounts of...