the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Oct 2021
| 06 Oct 2021
Authigenic rhabdophane from brown iron ore of the oxidation zone of the Babaryk massive sulfide occurrence (South Urals): scanning electron microscope (SEM) and electron backscattered diffraction (EBSD) study
Elena V. Belogub
Vladimir V. Shilovskikh
Konstantin A. Novoselov
Ivan A. Blinov
Ksenia A. Filippova
Rhabdophane (Ce0.34−0.43Nd0.13−0.14Ca0.06−0.29La0.08−0.11Y0.05−0.12Pr0.03−0.05Sm0.02−0.05Gd0.02−0.05Fe0−0.04Dy((P0.69−0.96S0.04−0.31)1.00O4)•H2O is found in a Fe3+-oxyhydroxide nodule (brown iron ore) collected from the upper part of the oxidation profile of the Babaryk massive sulfide occurrence (South Urals, Russia) at a 1.6 m depth. The structural and microtextural features of rhabdophane are revealed by electron backscattered diffraction (EBSD); the chemical composition and distribution of the main components are determined on a scanning electron microscope (SEM) equipped with an energy-dispersive analyzer (EDA); the bulk contents of rare earth elements (REEs) and other elements in rock samples are analyzed using inductively coupled plasma mass spectrometry (ICP-MS). Rhabdophane forms spherulitic aggregates up to 35 µm in size with a fine-grained core and radial radiant rims composed of prismatic crystals. The chaotically oriented aggregates of its particles of various sizes including prismatic crystals and spherulitic intergrowths also fill fractures up to 200 µm long and 20–30 µm thick in goethite. The zonal radial radiant structure of the rhabdophane aggregates and their occurrence in fractures of goethite unambiguously indicate the authigenic origin of rhabdophane. The chemically heterogeneous rhabdophane grains always contain Y, Ca and S and rarely Fe and Sr and are Th- or U-free. Contrasting zonation of Ca, S and Y contents is characteristic of spherulites. The band contrast of the EBSD patterns shows a good crystallinity of prismatic crystals regardless of the chemical composition even for Ca–S-rich zones. On the other hand, the Ca- and S-rich fine-grained centers of the spherulites do not yield any distinguishable diffraction patterns. There is a strong negative correlation in pairs (Ca+Sr)–P and (REEs+Y)–S and a positive correlation in pairs (Ca+Sr)–S and (REEs+Y)–P, which indicates the isomorphism according to the scheme (REEs+Y)3+ + (PO4)3− ↔ (Ca+Sr)2+ + (SO4)2−. Thus, the chemical composition of rhabdophane does not completely correspond to the rhabdophane–tristramite/brockite series because of the absence of tetravalent U or Th. In contrast to similar samples from the deeper part of the oxidation zone, the brown iron ore with rhabdophane is enriched in light rare earth elements (LREEs) and P. The REEs were probably sourced from ore-bearing volcanomictic rocks, while P could also have been derived from the soil. The enrichment in REEs and P and the formation of rhabdophane are related to the alternation of dry and wet periods, the P input, and sorption–desorption of REEs from Fe3+ oxyhydroxides and/or clay minerals due to pH changes and variable composition of pore water.
- Article
(18269 KB) - Full-text XML
-
Supplement
(948 KB) - BibTeX
- EndNote





Phosphates are the most common mineral forms of rare earth elements (REEs) in igneous and metamorphic rocks. As a rule, they include anhydrous phases: monazite, which concentrates light rare earth elements (LREEs), and xenotime, which concentrates Y and heavy rare earth elements (HREEs). In addition to simple anhydrous phosphates, florensite is also found in postdiagenetic Al-rich sedimentary rocks (Rasmussen et al., 1998). In the weathering crust of black shales and gold placers, a specific type of impurity-rich monazite (kularite) was found as spherical particles with inclusions of rock-forming minerals (Makeev et al., 2020). Rhabdophane, a hexagonal hydrous REE phosphate, mainly occurs in late hydrothermal associations of alkaline rocks (syenites, carbonatites) and polymetallic veins associated with granites (http://www.mindat.org, last access: 1 October 2021).
In unmetamorphosed sedimentary rocks, REE phosphates usually form very small grains; thus their precise identification is extremely difficult. As a rule, many findings are typically referred to as varieties of monazite. For example, monazite is mentioned in the ooidal ironstones of the Techa (South Urals) (Novoselov et al., 2018) and Bakchar (Western Siberia) (Rudmin et al., 2020) deposits. Rudmin et al. (2020) report on a deficit in the analytical total and a discrepancy between the electron backscattered diffraction (EBSD) patterns of the studied and standard monazite, but a conclusion about belonging to a specific mineral species is not drawn. An intermediate member of the rhabdophane–tristramite series was probably found in ooidal ironstone of the Kamysh-Burun deposit (Kerch Peninsula, Crimea) (Sokol et al., 2020). Phosphates are considered to be one of the main concentrators of REEs+Y in coals (Seredin and Dai, 2012, and references therein). This phosphate form can be stable and remains in the coal combustion byproducts (Montross et al., 2018). Secondary monazite and rhabdophane are mentioned amid REE minerals of the Kejal kaolin deposit (Iran) (Abedini et al., 2012) and the Alibaltalu bauxitic lateritic deposit (Azerbaijan–Iran) (Mikaeli et al., 2009) and weathering profile of granites of Madagascar (Berger et al., 2008, 2014) and Japan (Ichimura et al., 2020). Rhabdophane is also described among the epigenetic minerals of the Bayan Obo REE deposit (Smith et al., 2000) and some regolith-hosted REE deposits (Li et al., 2017). Most references, however, provide no comprehensive evidence in favor of determining the precise mineral species of REE phosphates. The recognition of hydrous and anhydrous REE phosphates is typically performed using the electron probe microanalysis (EPMA) based on the deficit of the analytical total (Scherer, 2000; Krenn and Finger, 2007; Montross et al., 2018) and rarely Raman spectroscopy (Škoda and Chopyakova, 2006; Berger et al., 2008, 2014).
The general formula of minerals of the rhabdophane group is (REE,Ca,Th)(PO4)•H2O (http://www.mindat.org, last access: 1 October 2021). Minerals of this group are crystallized in the space group P6222, excluding tristramite, for which the space group is not determined. A hexagonal structure of rhabdophane can be shown as an alternation of two types of layer parallel to plane (100). The first layer formed by REE atoms and phosphate tetrahedrons appears to be very close to monazite, while the second one comprises the chains formed by water molecules and REE and phosphates groups. This layer alternation leads to a looser packing in comparison with the monazite-type structure (Romero et al., 1994; Clavier et al., 2011). These features explain wide variations in water content of these REE phosphates (Shelyug et al., 2018), as well as their higher isomorphic capacity compared to that of monazite.
Calcium is dominant in two members of the rhabdophane group: brockite (Ca,Th,Ce)PO4•H2O and tristramite (Ca,U,Fe)(PO4,SO4)•2H2O. The entry of Ca2+ to the rhabdophane structure is suggested due to a heterovalent isomorphism according to the scheme 2REE Ca Th(U)4+ (brockite) or REE PO ↔ Ca SO (tristramite). Both mineral species include tetravalent cations for the compensation of Ca2+. Doynikova et al. (1993) described Ca-prevailing “smirnovskite” (Ca,REE)PO4•nH2O with the same space group, but later this mineral species was not confirmed. In this work, we studied a Ca- and S-rich U- and Th-free rhabdophane with a widely variable chemical composition from the upper part of the oxidation zone of the Babaryk volcanic-hosted massive sulfide (VHMS) occurrence (South Urals). The work aims to reveal the features of authigenic REE mineral forms in the brown iron ores, to clarify the isomorphic schemes of rhabdophane and to elucidate the reasons for its formation. To determine the type of the mineral structure and its chemical composition, we applied EBSD and a scanning electron microscope (SEM) with an energy-dispersive analyzer (EDA).
The oxidation zone of the Babaryk occurrence was studied in drill holes and trenches during prospecting works (Novoselov et al., 2009). During the detailed study of the oxidation zone, REE mineralization was found in borehole no. 5889. Eight samples of brown iron ore and limonite ocher were collected from various depths of the borehole. Some samples from trenches and other boreholes were also studied.
Polished sections of brown iron ores were monitored using an Axioscope A.1 CZ optical microscope and scanning electron microscopes (SEMs) at the Institute of Mineralogy (IMin), South Urals Federal Research Center of Mineralogy and Geoecology, Urals Branch of the Russian Academy of Sciences, Miass, Russia. The chemical composition of most representative rhabdophane grains was determined on a Tescan Vega 3 SBU SEM equipped with an Oxford Instruments X-act EDA at an accelerating voltage of 20 kV and a beam current of 200–300 nA in spot mode. The energy-dispersive (ED) spectra were acquired, processed and quantified in the Inca 5.02 software. The EDA detection limit is reported to be no worse than 0.3 wt %. The spectrometer was calibrated following the quant optimization procedure on pure Co before and after analysis, and the beam current drift was less than 1 %. The following standards were used for quantification: monazite for P, Ce, La, Pr, Nd and Sm; apatite for Ca; baryte for S; hematite (MINM25-53/01-044) for Fe; xenotime for Y and Dy; celestite (NERMA.GEO1.25.10.74GT) for Sr; and Gd3Ga5O12 (N 1362) for Gd.
The REEs and Y, Th, U and P contents of brown iron ore were determined on an Agilent 7700x inductively coupled plasma mass spectrometer equipped with a MassHunter software package and standards SGD-2a and ZUK at the IMin. The Ce anomaly value was calculated as , where Ce is CeN and Ce* is (LaN × PrN) (normalized to chondrite; Taylor and McLennan, 1985). The bulk mineral composition was determined using the Rietveld refinement method and Siroquant 4.0 software with an internal base of standards. The X-ray diffraction (XRD) patterns were obtained on a Shimadzu XRD-6000 (Cu-Kα radiation with a graphite monochromator, 1 ∘/min, interval 2Θ of 4–70∘).
Crystal chemical formulas of rhabdophane were recalculated to one cation atom in an anionic group. The water content was calculated from the stoichiometry of rhabdophane. The analytical total close to 100 % (taking into account the calculated water) was used as a criterion of analytical correctness.
Element distribution maps and additional data on chemical composition (Supplement file S2) including an element distribution profile through the rhabdophane particles were obtained at the GEOMODEL resource center (Saint Petersburg State University (SPbU), St Petersburg, Russia) on a Hitachi S-3400N SEM equipped with an Oxford Instruments X-Max 20 EDA at an accelerating voltage of 10 kV and a beam current of 1 nA to increase the locality of the ED analysis. The spectrometer was calibrated against a set of standard natural and synthetic samples, including REE-bearing glasses (Micro-Analysis Consultants Ltd (MAC) standards).
The EBSD maps were obtained using an Oxford Instruments HKL NordlysNano detector (GEOMODEL resource center, Scientific Park, SPbU). The acquisition conditions were as follows: 30 kV and 1.5 nA, exposition 0.5 s per pattern, averaging 5–10 images (when mapping) or 60 images (to obtain individual patterns), and their subsequent processing using the software package Oxford Instruments AZtecHKL. Several structural patterns of hexagonal rhabdophane from the Inorganic Crystal Structure Database (ICSD) and monoclinic monazite from the American Mineralogist Crystal Structure Database (AMCSD) were used as the comparison phases. The data on the orientation of the individual crystals in rhabdophane aggregates are shown as Euler color schemes, pole figures and orientation distribution density heatmaps (Mason and Schuh, 2009).
To obtain a mechanically undistorted surface, the sample for EBSD analysis was treated with a direct beam of Ar plasma using an Oxford Instruments Ionfab300 etcher, an exposition of 10 min, an angle of 45∘, an accelerating voltage 500 V, a current of 200 mA, and a beam diameter of 10 cm (Nanophotonics resource center, Scientific Park, SPbU).
The Babaryk VHMS occurrence is located 18 km NE of the town of Magnitogorsk, in the Aleksandrinka ore area (Chelyabinsk region, Russia), which hosts the eponymous polymetallic VHMS deposit. The Babaryk occurrence occurs in the northern part of the Magnitogorsk Zone of the Urals (Kozlov, 2001; Puchkov, 2017), which is composed of Devonian volcanic and volcanosedimentary rocks, as well as of syngenetic subvolcanic bodies.
The Babaryk occurrence consists of the following sequences (from bottom to top): (i) ore-bearing aphyric basalt, pillow lavas, dacite, rhyodacite and their lavoclastites of the Middle Devonian Aleksandrinka Formation; (ii) calcareous volcanomictic rocks of andesitic composition of the Middle Devonian Urlyady Formation with sulfide clasts at the basement; and (iii) siliceous siltstone, limestone and tuffite of the Upper Devonian Babaryk Formation (Fig. 1). The structure of the occurrence is complicated by faults, which led to the formation of a collapse zone located above the ore-hosting sequence with higher thickness of a weathering crust of up to 75 m.
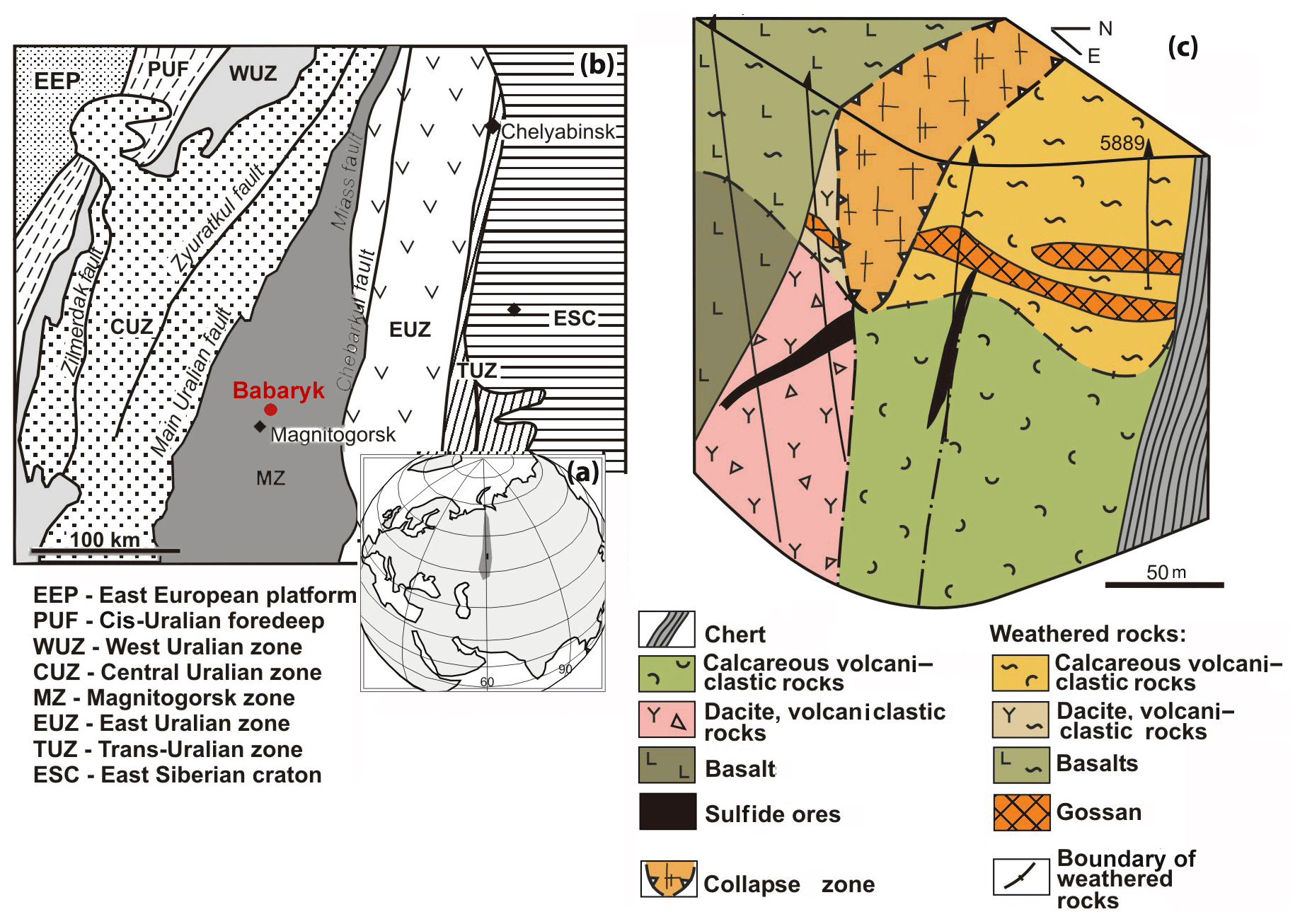
Figure 1Geographical (a) and geodynamic (b) setting (after Kozlov, 2001) and geological block diagram (c) of the Babaryk VHMS occurrence. Borehole no. 5889 is shown.
The Babaryk occurrence belongs to the Kuroko type of VHMS deposits grading 2.15 wt % Cu, 5.56 wt % Zn, 1.93 wt % Pb, 0.88 g/t Au and 108.6 g/t Ag (Novoselov et al., 2009). Primary massive sulfide ores form three lenticular bodies. The main ore body no. 1 up to 20 m thick dips to the west at an angle of 25–35∘ and is cut by a fault from the east (Fig. 1c). The host dacite and dacitic lavoclastites are altered to quartz–sericite schist at the bottom of the ore body. At the east, the ore body has contact with calcareous volcanomictic rocks along a fault zone. Ore body no. 2 of banded polymetallic ores is 1–2 m thick, occurs at the boundary with a gold-bearing oxidation zone and is hosted by pyritized chlorite and pyrite–quartz–sericite schist. Thin ore body no. 3 of stringer-disseminated fluorite-bearing sulfide–baryte ores is localized in the fault zone (Fig. 1c).
Pyrite, sphalerite, chalcopyrite and bornite are the major primary ore minerals. Galena, tetrahedrite and tennantite are minor, and native gold (electrum), germanite, colusite, argyrodite, renierite(?), Ag-bearing chalcocite, stromeyerite and a cervelleite-like sulfotelluride are rare. Gangue minerals include quartz, baryte, calcite, sericite and rare fluorite (Novoselov et al., 2006).
The weathering crust at the area of the ore occurrence consists of talus and oxidized residual sediments (eluvium). The talus sediments 7–11 m thick are unevenly distributed. The residual sediments occur below talus sediments. The oxidation zone develops mainly after disseminated ores in volcanic and volcanosedimentary host rocks. The collapse zone with a maximum thickness of the oxidation zone of up to 75 m is located above the leached massive sulfide ores of ore body no. 2. The oxidation zone has a typical cross-section including (from top to bottom) an “iron hat”, a thin leaching subzone and a cementation subzone. The iron hat occurs under a thin soil horizon 0.3–0.5 m thick and mainly consists of clayey–limonite ocher with inclusions of denser Fe3+-oxyhydroxide brown iron ore with jarosite veins and aluminite veins up to 2–3 cm thick in the lower part. The quartz–baryte and pyrite sands occur between the iron hat zone and primary massive sulfide ores. The pore waters circulating in the sands were saturated with water-soluble Zn and Mg sulfates, which formed when the drill core dried up. Small interlayers of native sulfur and/or gypsum occurred in sandy layers of some boreholes. The cementation subzone contains covellite and chalcocite, which replace primary sulfides (Novoselov et al., 2006).
The weathering crust and oxidation zone formed under tropical peneplain conditions starting from the Triassic and were followed by semiarid conditions at the beginning of the Cenozoic, which are still characteristic of this region (Sigov, 1969). Because of tectonics and variegated composition of the host rocks, the weathering profile cannot directly be compared with that of the undisturbed rocks as described by Berger et al. (2008), Janots et al. (2015), Li et al. (2017), Fu et al. (2019) and Ichimura et al. (2020).
4.1 Structure and mineral and chemical compositions of the brown iron ores
Borehole no. 5889 was drilled 50–60 m northeast of the main ore body to cross the gold-bearing oxidation zone. Under a 30 cm soil horizon, there are talus clayey sediments extending to a depth of 11 m and containing small nodules of dense Fe3+-oxyhydroxide brown ore with veinlets and films of Mn oxyhydroxides. Rhabdophane was found only in one sample from a depth of 1.6 m. The main body of the residual brown iron ore represented by alternating dense, ocher, and ocher–clayey varieties occurs at a depth of 11–35 m. The structural clayey residual sediments after volcanomictic sandstones are observed deeper than 35 m. Goethite and quartz are the major minerals of the brown iron ore. Some samples contain minor kaolinite and hydrous mica. A significant quantity of Mn oxyhydroxides is present in the brown iron ore at a depth of 25.2 m. The deepest sample contains minor calcite (Table 1, Supplement file S1). The proportions between minerals vary. The half width at half maximum (HWHM) of the main reflection of goethite (4.18 Å), reflecting the perfection of its structure, changes irregularly with depth (Table 1).
Table 1Mineral composition of the brown iron ore from borehole no. 5889 according to XRD analysis (wt %).
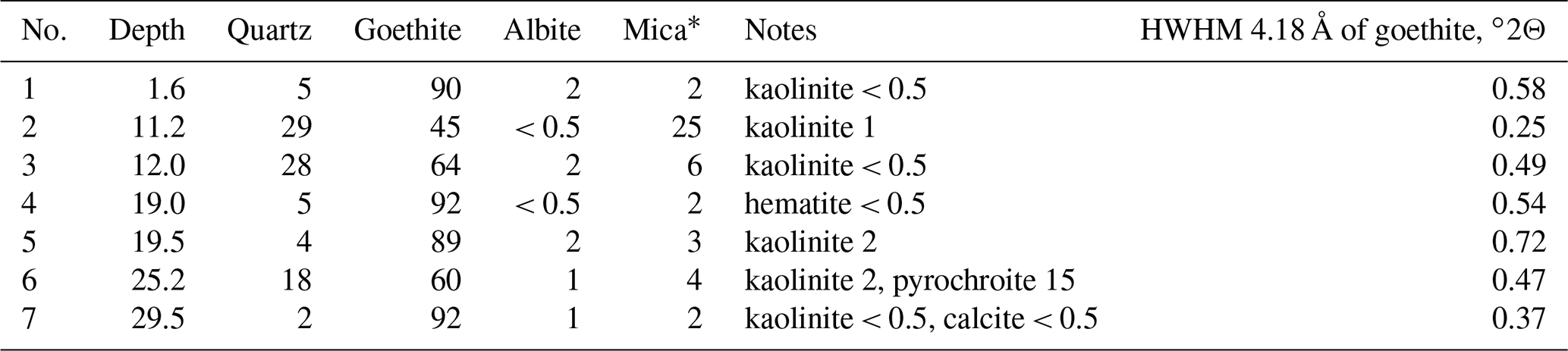
Note that mica is hydrous because of the asymmetrical shape of the first basal reflection (see XRD patterns, Supplement file S1).
The bulk chemical composition of samples significantly varies with respect to P, U, Th, Y and ΣREE contents. The maximum amount of P (1.3 wt %), U (15.9 ppm) and ΣREEs (446 ppm) is determined in a rhabdophane-bearing sample taken at a depth of 1.6 m. This level can be considered an analog of zone B (illuvial) of a typical weathering profile (Janots et al., 2015; Li et al., 2017, and references therein). This sample has the higher contents of P (by 3 or more times) and U and ΣREEs (by 2 or more times) in comparison with deeper brown iron ore samples. In the precursor calcareous volcanomictic sediments, the P content is only 0.03 wt % and ΣREE content is 31.19 ppm. Relative to the precursor calcareous volcanomictic sediments, the brown iron ores are significantly enriched in P and, to a lesser extent, in REEs, U and Y (Table 2).
Table 2Content of P (wt %), Y, REEs, U and Th (ppm) of the brown iron ore and possible precursor calcareous volcanomictic rock (89 m).
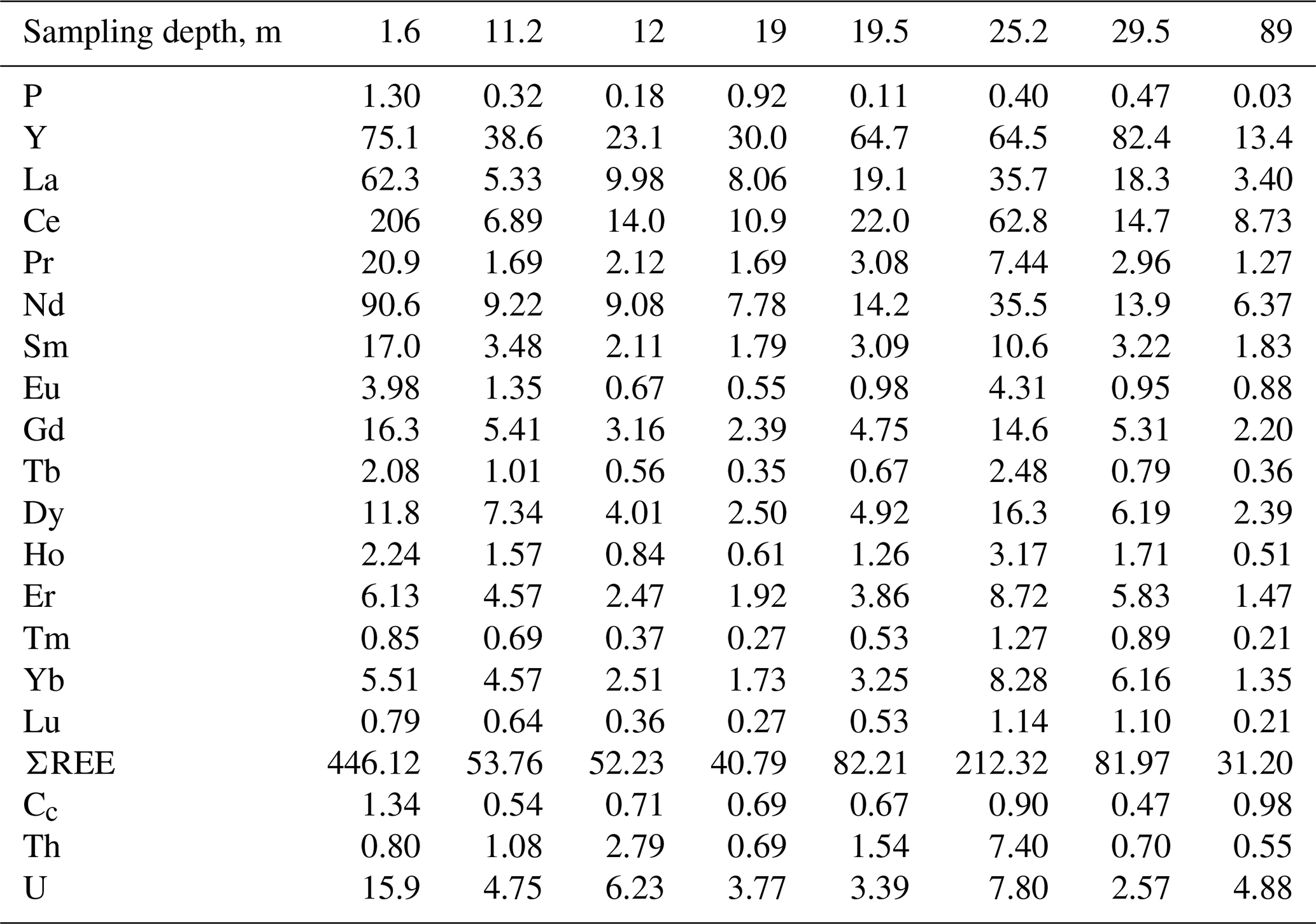
Note that Cc is the cerium anomaly.
The sample with rhabdophane is a rounded nodule 2.5 × 3 × 3.5 cm in size (Fig. 2a), which consists of fragments of ferruginous silty sandstones up to 10 mm in size, and is enclosed in Fe3+ oxyhydroxides with minor quartz and clay minerals (Fig. 2b). A crust of dense colloform goethite and Mn oxyhydroxides up to 0.15 mm thick has developed along the periphery of fragments of silty sandstone (Fig. 2c, d). Porous radial aggregates of acicular goethite occur in the cavities between the fragments of silty sandstone (Fig. 2e, f).

Figure 2Brown iron ore nodule, sample 5889/1.6. (a) General view; (b) clastic structure of brown iron ore; (c, d) fragment of psephitic ferruginous rocks with crusts of compact Fe and Mn oxyhydroxides; (e, f) radial radiant aggregate of acicular goethite with interstitial clay minerals between fragments of ferruginous rocks (b – oblique light; c–f – reflected light; c, e – with analyzer; d, f – without analyzer).
4.2 Morphology of rhabdophane
The rhabdophane aggregates form two morphological varieties. The first-type aggregates reach a maximum of 200 µm in length (Fig. 3a–c) and consist of chaotically oriented lamellar crystals ranging from a few hundreds of nanometers to 10 µm in size (Fig. 3b) and, locally, small spherulitic intergrowths with a very fine grained central core (Fig. 3c). These aggregates are located among colloform crusts of Fe3+ oxyhydroxides filling the cavities or fractures; thus their growth was spatially limited. The second-type spherical radial radiant aggregates up to 35 µm in size exhibit a displaced spherical core composed of nano-scale individuals and asymmetric zonal elongated prismatic crystals 0.5–2.0 µm thick at the periphery (Fig. 3d, f). These aggregates occur among acicular goethite, and they apparently did not experience growth restrictions.

Figure 3Rhabdophane aggregates in the brown iron ore: (a) zonal fine-grained, (b) zonal with fine-grained and radial radiant filling of the central cavity; (c) fine-grained with four spherical growth centers with radial rims; (d) zonal spherule; (e, f) spherical radial radiant intergrowths with displaced fine-grained center.
4.3 Chemical composition of rhabdophane
The composition of rhabdophane is shown in Table 3 and Supplement file S2. Table 3 shows only Tescan Vega 3 analyses supported by a full set of international standards. Supplement file S2 includes all chemical analyses.
Table 3Representative chemical composition (wt %) and crystal chemical formulas of rhabdophane.
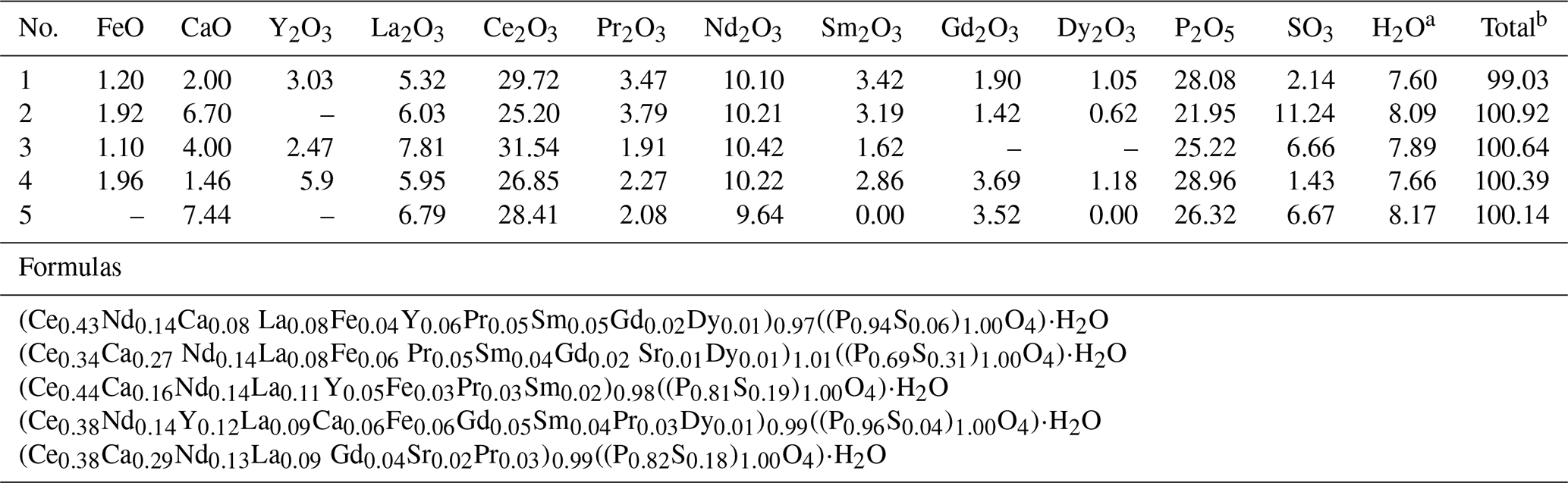
Notes that SrO content in analysis 2 is 0.56 wt % and in analysis 5 is 1.09 wt %. a Calculated from stoichiometry of rhabdophane (including one H2O molecule). b With calculated H2O content.
Table 4Paired correlation coefficients of main elements of rhabdophane.
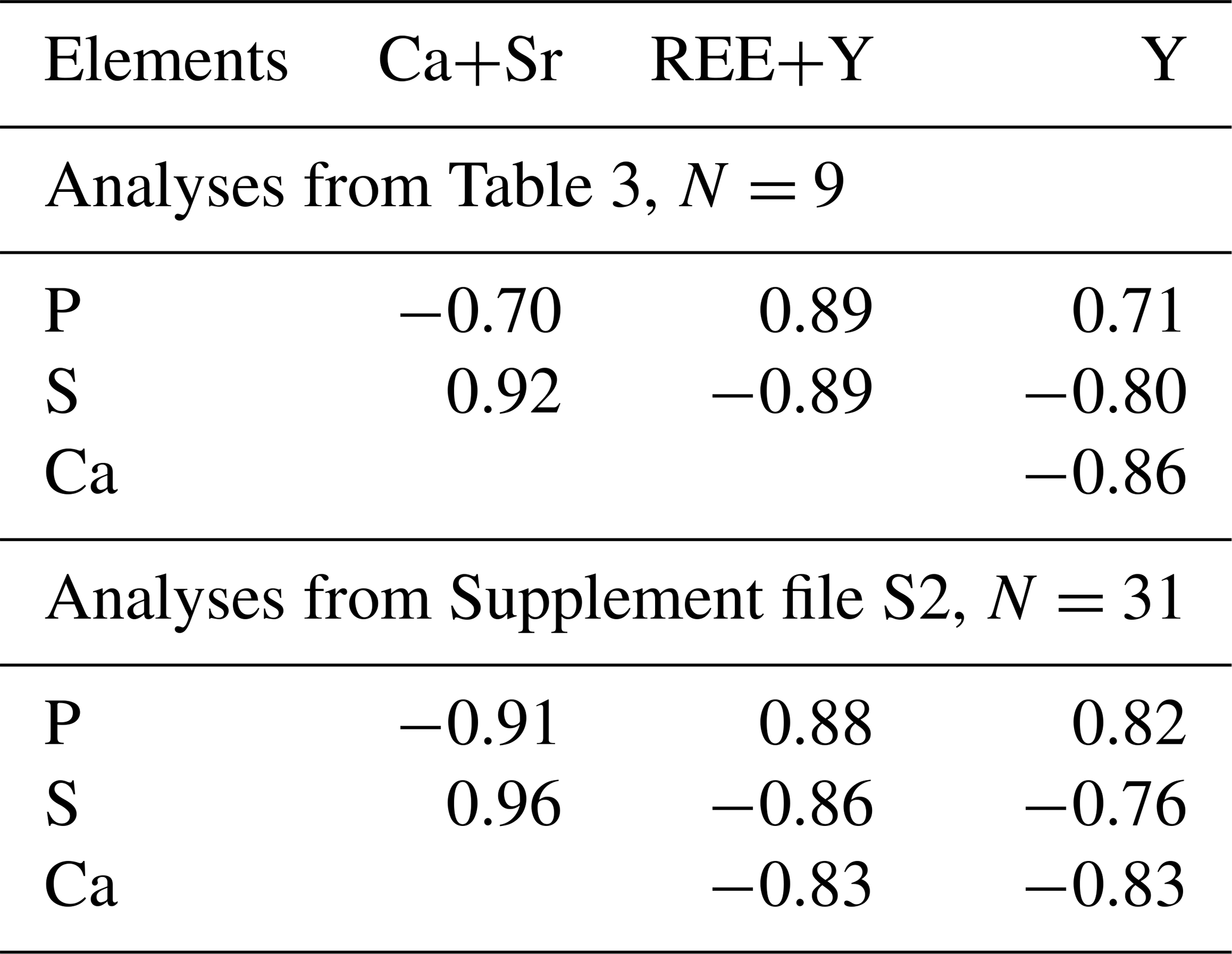
Cerium, Nd and Ca are the dominant cations of rhabdophane. It also contains Sr, Fe and S. The presence of Al is detected in some spectra. The dominant anion-building elements include P and S. The representative analyses yield rather correct formulas according to the cation anion ratio. Taking into account the calculated H2O content (per 1 molecule, as in the formula of rhabdophane), the analytical sum is close to 100 %. According to formal criteria, all analyses correspond to rhabdophane-(Ce). The Ce content varies from 0.34 to 0.44 atoms per formula unit (a.p.f.u.); the Ca contents range from 0.06 to 0.29 a.p.f.u.; and more consistent Nd contents vary from 0.13 to 0.14 a.p.f.u. (Table 3).
Rhabdophane aggregates are chemically heterogeneous. A profile through a spherical aggregate (Fig. 4), as well as the binary diagrams (Fig. 5), shows that (Ca+Sr) and S, as well as (Y+REE) and P, have a high positive correlation (Table 4), whereas a high negative correlation is observed between Y, REE and S, and Ca with P (Fig. 5, Table 4). The slope of the trend lines is also the same for the populations of analytical points of different SEMs.
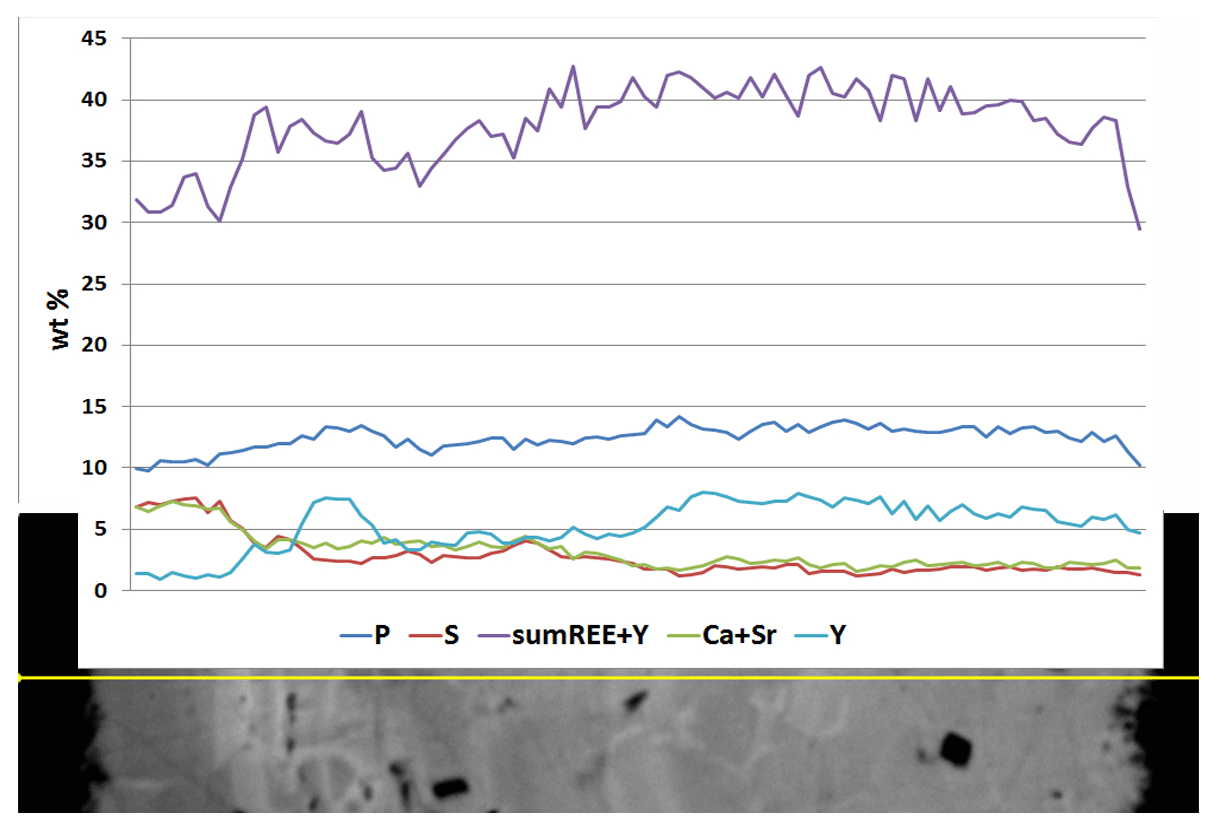
Microgeochemical maps of rhabdophane aggregates show a relatively uniform distribution of P and Ce in contrast to Ca, Y and S (Figs. 6–8). The fine-grained cores of spherical aggregates are enriched in Ca, Sr and S, whereas the radial radiant rims of spheres are typically enriched in Y (Figs. 6, 8). There are also spherical aggregates with reverse chemical zoning (Fig. 7). The distribution of the main elements in fine-grained aggregates intergrown with elongated crystals is more complicated (Fig. 8).
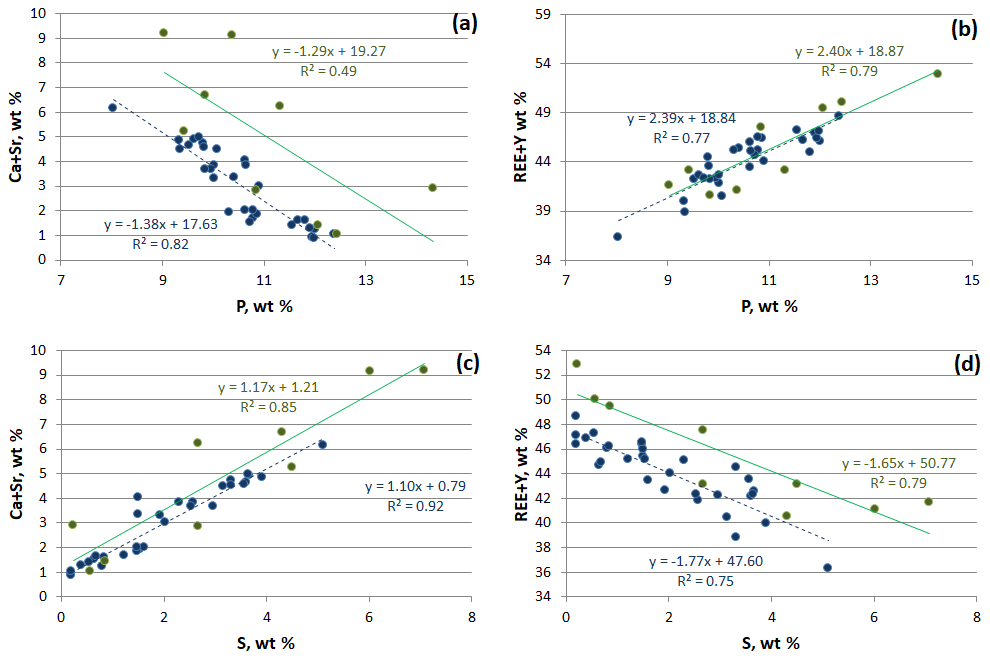
Figure 5Correlation between main elements of rhabdophane. Green, Tescan Vega 3 SEM analyses; blue, Hitachi S-3400N SEM analysis.
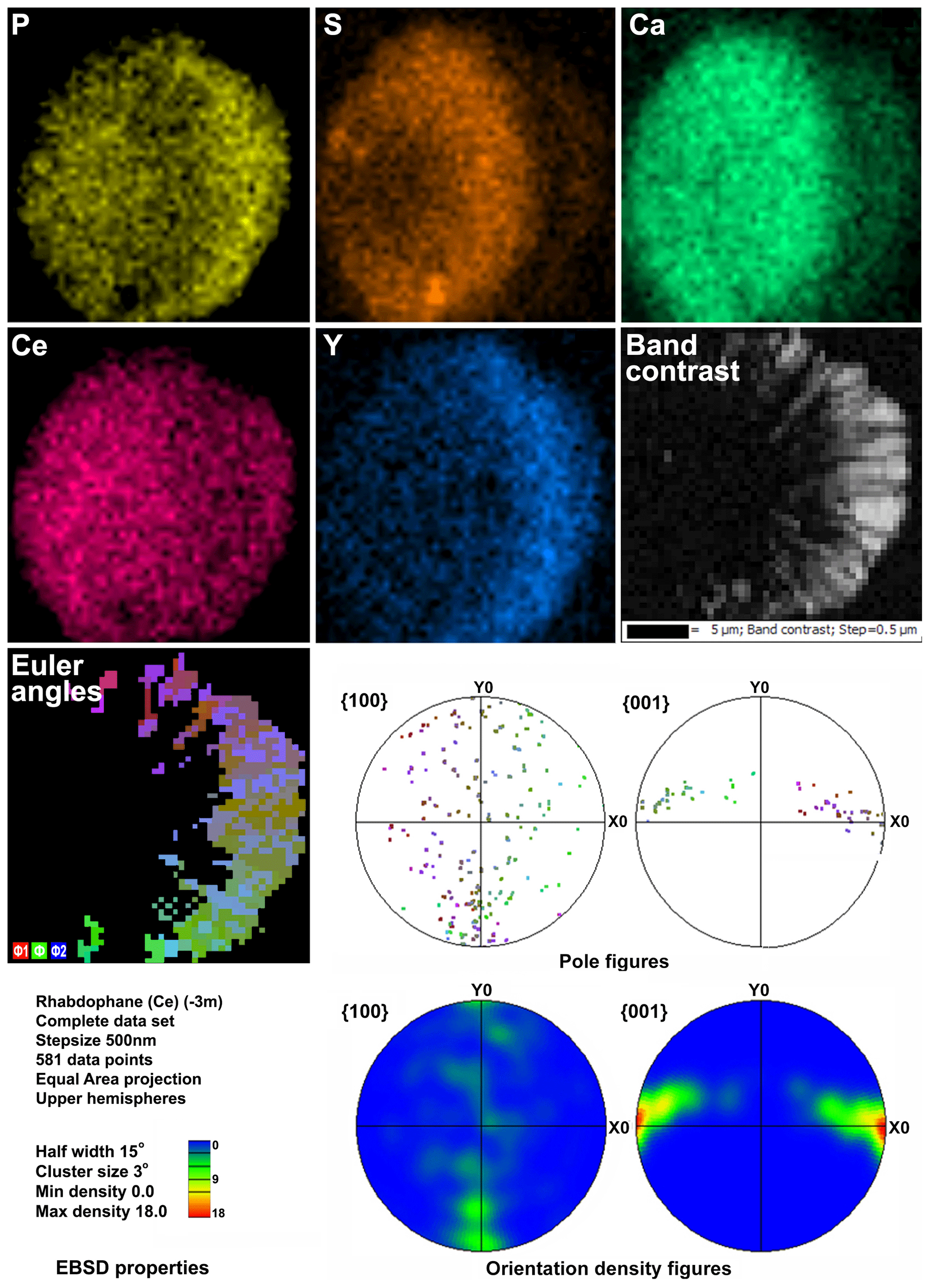
Figure 6Distribution of elements, EBSD band contrast, rhabdophane crystal orientation in Euler colors, pole figures (each point in the pole figures corresponds to one point in the rhabdophane crystal orientation map and retains its color) and orientation distribution density heatmaps of spherical rhabdophane aggregate; for BSE image, see Fig. 3e.
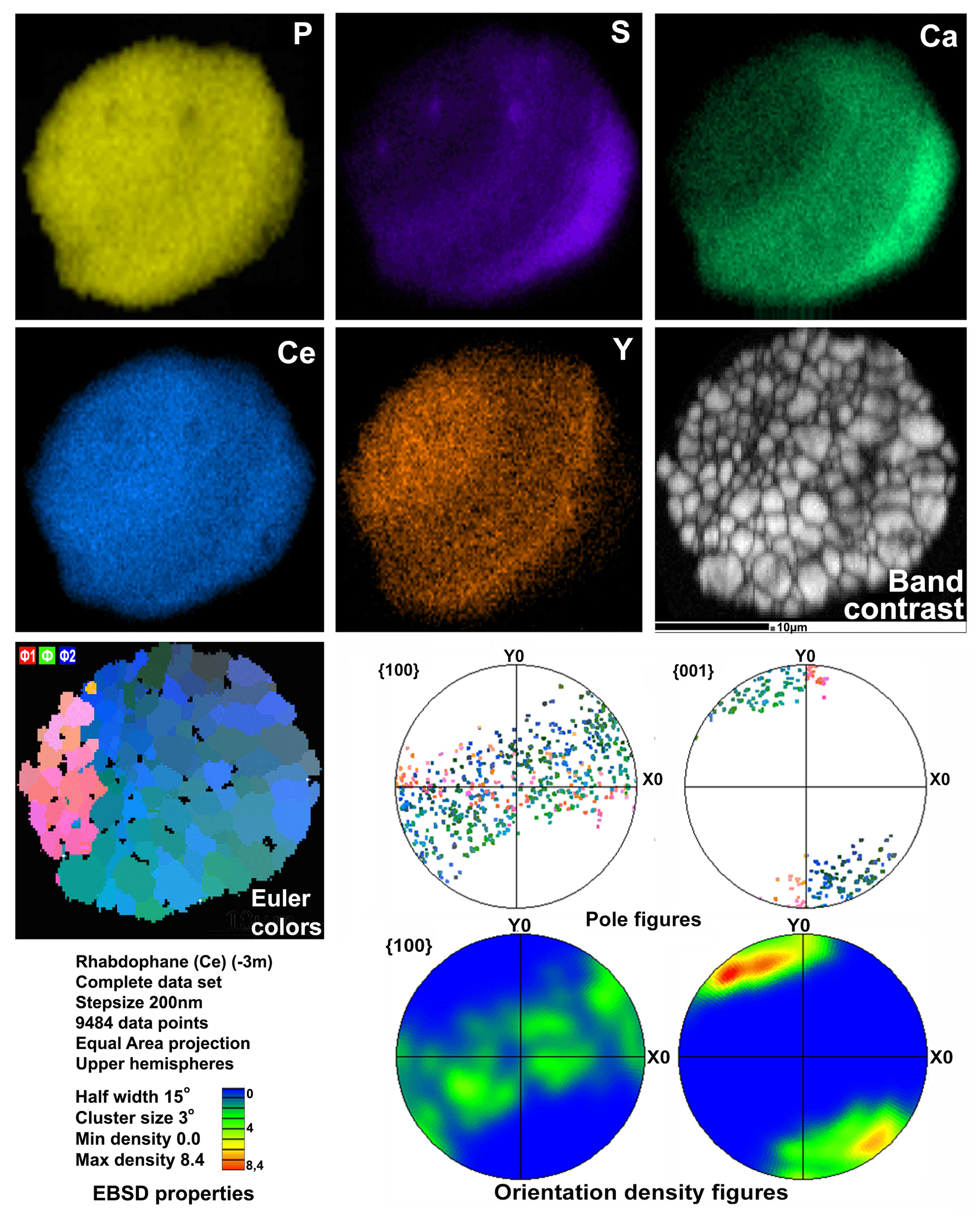
Figure 7Distribution of elements, EBSD band contrast, rhabdophane crystal orientation in Euler colors, pole figures (each point in the pole figures corresponds to one point in the rhabdophane crystal orientation map and retains its color) and orientation distribution density heatmaps of spherical rhabdophane aggregate; for BSE image, see Fig. 3d.
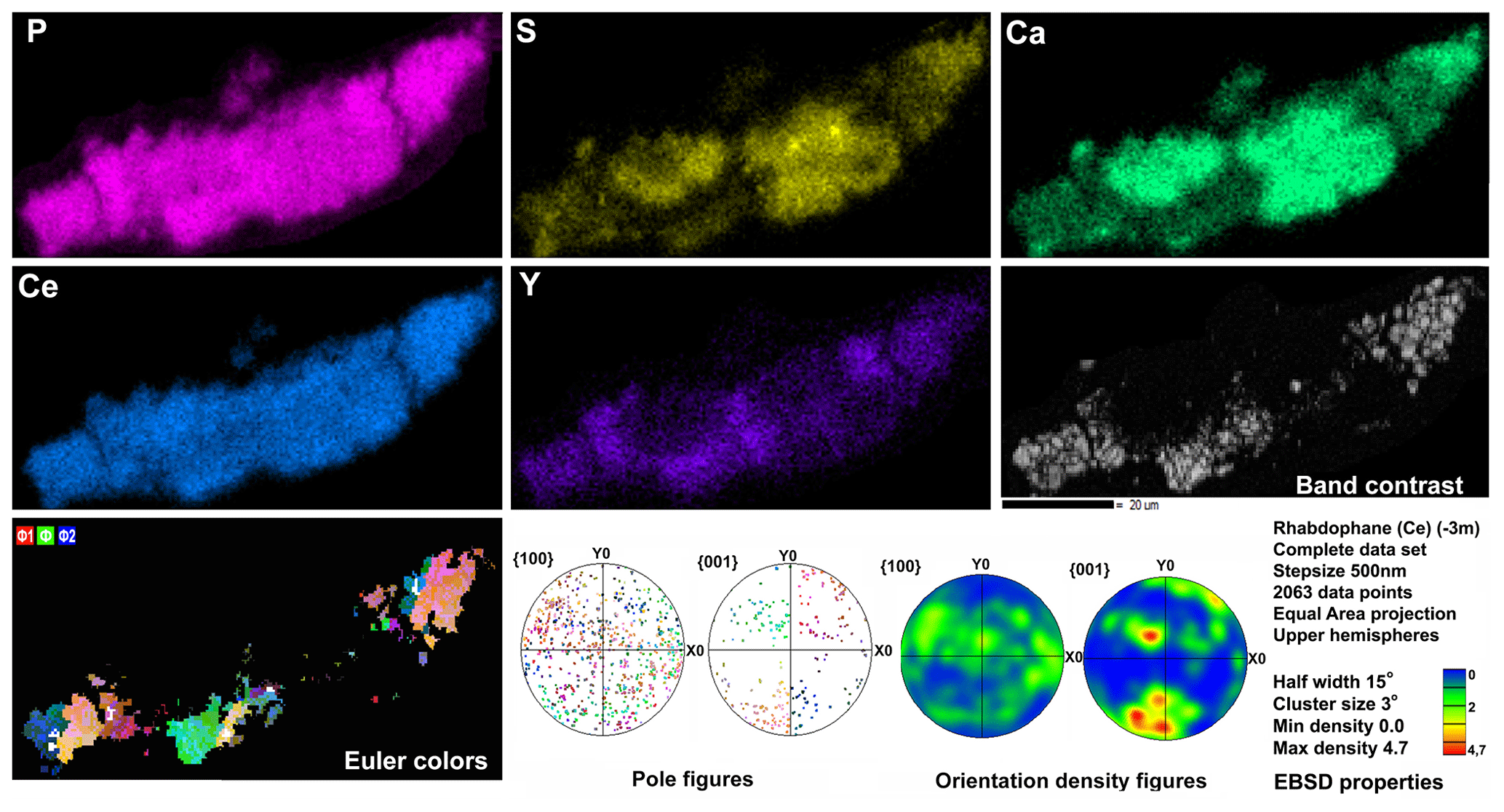
Figure 8Distribution of elements, EBSD band contrast, rhabdophane crystal orientation in Euler colors. pole figures (each point in the pole figures corresponds to one point in the rhabdophane crystal orientation map and retains its color) and orientation distribution density heatmaps of rhabdophane aggregate with four spherical growth centers; for BSE image, see Fig. 3c.
4.4 EBSD results
The Kikuchi patterns of the studied phosphates correspond to the reference hexagonal rhabdophane (ICSD 27859) at the coincidence level of 12 bands out of 12 with a mean angle deviation (MAD) of less than 1∘ (minimum 0.27∘) (Fig. 9). The best Kikuchi patterns were obtained from crystals that form rims around spherical aggregates. At the same time, even smaller crystals from fractures in goethite demonstrate fairly good crystallinity (Fig. 9a): when comparing Kikuchi patterns with the standard, 9–12 bands correspond to standard rhabdophane. One of the largest prismatic crystals exhibited the best Kikuchi pattern (Fig. 9b). The centrifugal growth is typical of spherical aggregates, while all crystallites starting from the nuclei are oriented regularly (Figs. 6, 7).
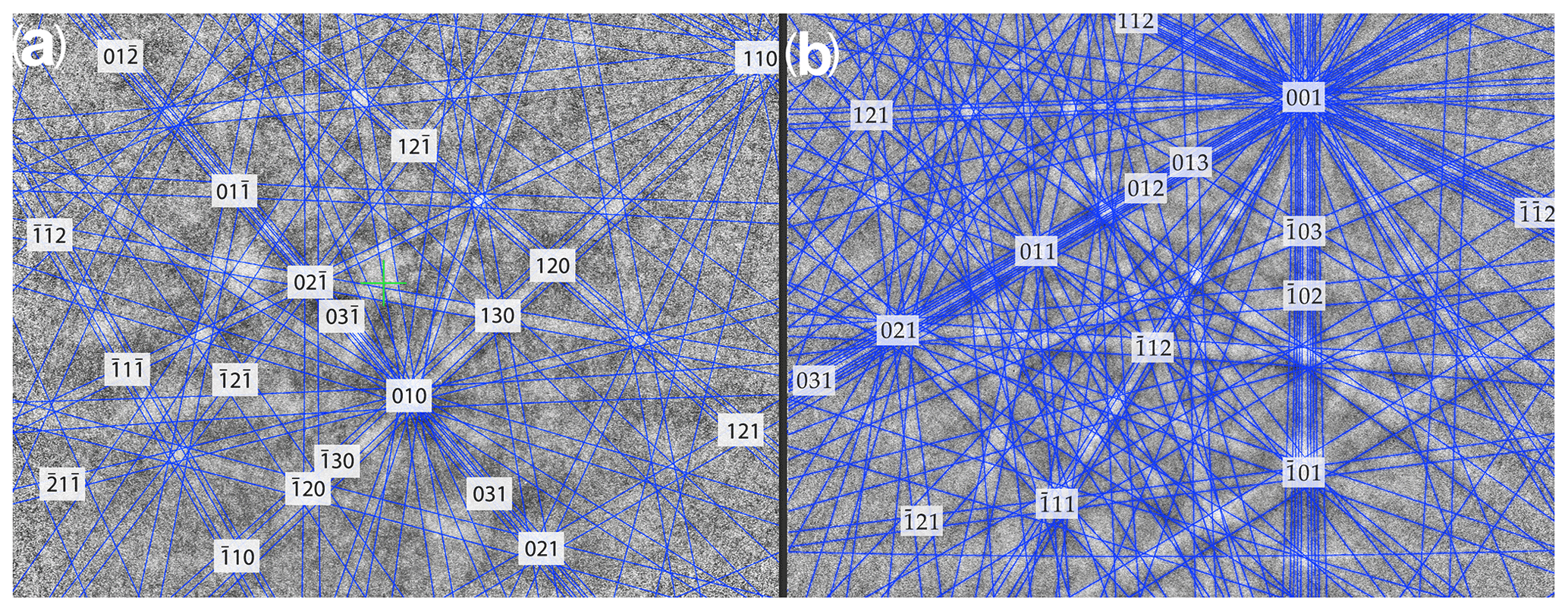
Figure 9Kikuchi patterns of rhabdophane from a fine-grained aggregate in the fracture of goethite (a) and prismatic crystal from the rims of radial radiant aggregate (b).
The degree of structural perfection of the studied rhabdophane aggregates varies. According to the band contrast patterns, the cores of spherical aggregates yield no EBSD patterns (Figs. 6, 8), while the later rims with evident zonal crystals exhibit a high band contrast (Figs. 6, 7). The areas of coherent scattering have different shapes and sizes of 0.5–2.5 µm and can be arranged in a mosaic manner (Fig. 7) or in the form of radial aggregates (Fig. 6). In this case, the band contrast patterns are sporadically correlated with the grain configuration in backscattered electron (BSE) images (Figs. 3, 6–8). For example, the particle from Fig. 3d in the BSE image is zonal due to various distributions of REEs, P, S and Ca. At the band contrast pattern, which is highly sensitive to the grain borders (Fig. 7), the aggregate has a mosaic structure and zoning is almost not traced in terms of the band contrast. In the studied section, the particle consists of the rounded domains ranging from 0.2 to 2.5 µm in size, which have different orientations (see the pattern in Euler's colors and pole figures, Fig. 7) with a tendency towards a radial arrangement of the crystallographic z axis and rotation of the x axis of individual crystals, similarly to the structure of the rim of the spherule shown in Figs. 3e, 3b and 6. In pole figures (Fig. 7), the angle between two areas of z-axis projections is 140–160∘. Projections of the x axis form a wide belt perpendicular to the z axis. This pattern corresponds to a section of the rim, similarly to that shown in Figs. 3e and 6. It is important that all domains have a comparably good crystallinity regardless of their composition (Fig. 7).
5.1 Morphological features
The studied rhabdophane forms elongated prismatic crystals typical of this mineral and is often assembled in radiant intergrowths and spherical aggregates. Its position in cavities of Fe3+ oxyhydroxides unambiguously indicates an authigenic origin. The rhabdophane aggregates formed later than Fe3+ oxyhydroxides in contrast to coprecipitation of rhabdophane with Fe3+ oxyhydroxides in the ooidal ironstone of the Kamysh-Burun deposit in Crimea (Sokol et al., 2020).
Early authigenic minerals of the rhabdophane group are described in saprolites of the laterite weathering crust developed after granites. In this case, however, rhabdophane either is pseudomorphic after monazite with relics of the latter or forms thin veins in sapropel (Berger et al., 2008, 2014). Rhabdophane–tristramite from the ooidal ironstone of the Kamysh-Burun deposit forms minute crystals within allochems or filled microfractures in them (Sokol et al., 2020).
Theoretically, spherulites 35–40 µm in size could be formed by coagulation and coalescence of colloidal particles. The presence of striking microcrystallites, however, contradicts this style of formation without subsequent recrystallization. At the same time, thermodynamic data indicate that the recrystallization of rhabdophane, which is metastable under ambient pressure, should lead to a transition into anhydrous monoclinic monazite, which is stable under weathering-crust conditions (Mooney, 1948; Shelyug et al., 2018). There is also no evidence of recrystallization of zonal rhabdophane aggregates in cavities of the colloform brown iron ore; thus, rhabdophane should crystallize from true solutions.
5.2 Chemical composition of rhabdophane
Rhabdophane from the Babaryk occurrence contains up to 4.5 wt % S (11.24 wt % SO3) and up to 5.32 wt % Ca (7.44 wt % CaO). Since the mineral is found in the oxidation zone of VHMS ores, the presence of S in its composition is expected. Apparently, these trace elements are typical of rhabdophane, which formed in the weathering crust, because rhabdophane formed after Madagascar granites also contains up to 9 wt % S and 7 wt % Ca and exhibits their heterogeneous distribution (Berger et al., 2014), and Kamysh-Burun ooidal ironstone, the REE phosphates of which also contain Ca (Sokol et al., 2020). In these localities, as well as in the Babaryk occurrence, Ca and S in rhabdophane are positively correlated. Berger et al. (2014) consider that they studied possibly a mixture of rhabdophane and tristramite. In our case, the double amount of crystallization water in tristramite in comparison with rhabdophane should result in a deficit of the analytical total calculated taking into account one H2O molecule, but this is not observed. Unfortunately, the lack of data on the space group of tristramite prevents the EBSD-based identification of this mineral. It should be noted that “kularite” (a variety of monazite) also contains Ca but by an order of magnitude less than in rhabdophane (Makeev et al., 2020).
5.3 Influence of composition on structure of rhabdophane and possible isomorphism schemes
Two rhabdophane spherulites exhibit different chemical zoning (Figs. 6, 7). The fine-grained center of one spherulite and the radial radiant rim of another spherulite are enriched in Ca and S. The fine-grained core and similar core of spherulites from the fracture in goethite yielded no Kikuchi patterns (Figs. 6, 8), whereas the crystalline rim exhibits a high band contrast (Fig. 7). Because of the similar composition of these parts of the aggregates, we believe that the low quality or absence of electron diffraction of the fine-grained core is related to the smaller size of the coherent scattering area rather than to the composition of the mineral. Thus, we suggest that Ca and S are incorporated into the crystal structure according to the isomorphism scheme (Y+REEs) (PO4)3− ↔ (Ca+Sr) (SO4)2−.
Some attempts have been made to synthesize the Ca-doped REE monazite-like phosphates (Gallini et al., 2005a, b; Sivakumar and Varadaraju, 2005), and they were successful if Ca2+ was coupled with U4+ or Th4+ (Bregiroux et al., 2007). The full substitution leads to the formation of minerals of the cheralite family with a monazite-like structure (Hugues et al., 1995; Clavier et al., 2011). There is evidence of the possible compensatory entry of Ca2+ with Ce4+ in the structure of monazite (Orlova et al., 2001, 2002; Kitaev et al., 2004). Taking into account similar structures of monazite and rhabdophane, we can suggest the same mechanism in our case. The possible incorporation of significant Ce4+ concentrations into phosphate minerals alternatively to cerianite and oxyhydroxides was indicated by Janots et al. (2015). Although no cerianite was found in our sample, the presence of Ce4+ in the structure of rhabdophane for the additional compensation of Ca2+ according to the scheme (Y+REEs)3+ + (PO (Ca+Sr)2+ + Ce4+ cannot be excluded because the sampling depth of 1.6 m implies access of free oxygen to the mineral-forming solution.
5.4 REE and P sources and formation of rhabdophane
According to the chemical composition, the brown iron ore sample with rhabdophane is characterized by significant enrichment in LREEs and the presence of a striking positive Ce anomaly in comparison with deeper brown iron ore samples, which exhibit an HREE enrichment and negative Ce anomaly. This is especially evident from a comparison of REE patterns of samples 5889/1.6 and 5889/11.2 (Fig. 10). The latter sample shows the lower ΣREE content, HREE enrichment and a negative Ce anomaly. The form of the REE pattern of sample 5889/1.6 is consistent with the Ce–Nd type of rhabdophane, which is the only REE-bearing mineral found in the brown iron ore sample from a depth of 1.6 m. The difference in the REE pattern of the sample taken from a depth of 25.2 m can be explained by a significant amount of Mn oxyhydroxide, which is subjected to higher HREE contents (Vereshchagin et al., 2020).
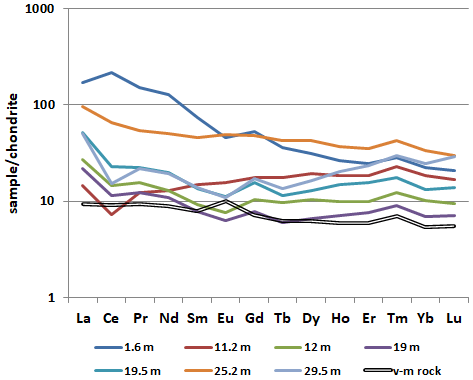
Figure 10Chondrite-normalized (Taylor and McLennan, 1985) REE pattern of the brown iron ore and volcanomictic rocks (v-m rocks).
The precursor calcareous volcanomictic rocks are characterized by a generally monotonic REE pattern with a minor Eu anomaly. The sorption of REEs on Fe3+ oxyhydroxides, which are the products of the formation of the oxidation zone, is the main reason for early REE scavenging from solutions with pH > 5 (Leyborne et al., 2006; Dinali et al., 2019). Almost all REE patterns, excluding patterns of samples from depths of 11.2 and 25.2 m, demonstrate a positive Gd anomaly, which is probably related to a negative Eu anomaly rather than to Gd accumulation.
The behavior of Ce in the weathering profile differs from that of other REEs because of its variable oxidation state: Ce4+ is less mobile than Ce3+. For example, strong Ce anomalies are characteristic of the upper part (illuvial horizon B) of the weathering profile of granites (Berger et al., 2008; Janots et al., 2015; Ichimura et al., 2020), as well as of regolith-hosted REE deposits (Li et al., 2017; Fu et al., 2019). The most likely reason for Ce accumulation in this horizon is the oxidation of Ce 3+ to Ce4+ and removal of Ce from soil solutions during precipitation of cerianite (Li et al., 2017; Ichimura et al., 2020). Oxidation of Ce is also possible with the participation of bacteria (Moffet, 1990), which can live in the soil pore water. Despite our weathering profile characterizing the tectonized rocks, we can compare a Ce-rich horizon with illuvial horizon B. According to Li et al. (2017), the Ce anomaly is a result of vertical pH and redox gradients in the weathering crusts, which facilitate the dissolution of REE-bearing minerals at shallow depths and fixation of REEs at a greater depth by either adsorption on clay minerals or precipitation of secondary minerals. Rhabdophane is the only REE-bearing mineral in our case. The amount of clay minerals in the brown iron ores is low, but the kaolinite content increases with the depth. The Fe3+ oxyhydroxides can act as effective sorbent; thus resorption of REEs from clay and Fe3+ oxyhydroxides can lead to the formation of rhabdophane. Previous studies of REE phosphates have shown that rhabdophane has lower thermodynamic stability and in the long term tends towards irreversible recrystallization into monazite with the loss of water molecule and an increase in the density of the structure. However, rhabdophane forms preferably in water solutions due to faster formation kinetics (Shelyug et al., 2018). A fast growth rate allows rhabdophane to be an effective REE scavenger from the pore solution.
Sorption–desorption processes involving REEs and P are caused by the properties of the sorbent surface (Borst et al., 2020) and the concentration of sorbate in the solution (Hughes and le Mare, 1982). The following processes of desorption of REEs from Fe3+-hydroxides can be considered: (i) the recrystallization of the sorbent with a decreasing specific surface area, (ii) the reduction of Fe3+ to Fe2+, (iii) the change in pH (Dinali et al., 2019, and references therein), (iv) the decrease in REE concentration of the solution and (v) the substitution of REEs with other cations in the absorbed complex.
In spite of the presence of the crystalline acicular goethite in our sample, the HWHM of its main reflection on XRD patterns indicates its strongly defective crystal structure. At the same time, the samples taken from the deeper horizons show high variability in the HWHM, which, in several cases, indicates a higher degree of perfection of the goethite structure, i.e., its significant recrystallization (Table 1). Therefore, the contribution of recrystallization to the enrichment of pore solutions in REEs cannot be ruled out. No evidence of Fe3+ reduction was found in our samples. Therefore, desorption due to changes in Eh proposed for the Kamysh-Burun ooidal ironstone (Sokol et al., 2020) is not applicable for the formation of the Babaryk rhabdophane.
A decrease in REE concentrations of the solutions is accompanied by the formation of REE minerals and may lead to further REE desorption from Fe3+ oxyhydroxides, which is supported by experimental data on possible formation of rhabdophane-(La) by the interaction of La-rich bentonite and P-rich waters (Szopa et al., 2012).
The changes in pH and REE concentrations of pore waters due to the alternating wet and dry periods probably significantly affected the sorption–desorption processes and REE migration. During wet periods, pore waters were diluted, pH increased and Eh decreased. During dry periods, pH became more acidic due to the oxidation of relict sulfides and ascending acidic capillary waters, and Eh and the concentration of solutions increased. The REE fractionation with redistribution of redox-dependent Ce in the weathering profile in borehole no. 5889 is possibly related to both periodic humidification and capillary ascending pore waters enriched in more mobile LREEs as a result of repeated sorption–desorption processes.
The fractionation of REEs between various authigenic minerals in supergene processes was previously shown on an example of U sandstone deposits of northern Bohemia (Scharmova and Scharm, 1994). Berger et al. (2014) also point to the REE fractionation between minerals during the formation of the weathering profile. These authors relate the fractionation to a different behavior of redox-sensitive Ce and indifference to oxidation REEs, as well as to distinct mobility of LREEs and HREEs at low temperatures. The fractionation and formation of REE minerals in ooidal ironstones are associated with a change in redox potential (Sokol et al., 2020).
It is suggested that the calcareous volcanosedimentary sediments of the Babaryk occurrence are the REE source. During oxidation in the presence of acidic sulfate waters, hyaloclasts and plagioclase in fragments of andesitic lavas become unstable and are transformed into a mixture of quartz and/or amorphous silica, hydromica, hydrochlorites and kaolinite. In this case, REEs are released to the solution and migrate or are adsorbed on clay minerals and Fe3+ oxyhydroxides (Ling and Liu, 2002). The formation of aluminite in the lower part of the iron hat, the source of Al for which plagioclase and/or hyaloclastic material was unstable under weathering conditions, is in agreement with a volcanogenic sedimentary source of REEs. An additional REE source can be related to a near-surface soil layer from which the REEs are transported to the lower levels of the weathering crust according to Berger et al. (2014) and Borst et al. (2020).
The formation of phosphates in the oxidation zone of ore deposits is a result of weathering of host rocks. Phosphorus and several other components of rocks enter the waters in the oxidation zone and participate in authigenic phosphate formation (Yakhontova and Grudev, 1987). For VHMS deposits, this is generally atypical. Nevertheless, the Pb sulfates–phosphates were found in the oxidation zone of the Aleksandrinka VHMS deposit located 5 km from Babaryk in a similar geological setting (Belogub, 1998).
The source of P in the near-surface brown iron ore can also be related to a soil–vegetation layer (Szopa et al., 2012) and an absorbed complex of Fe3+ oxyhydroxides. It was shown in an example of ferromanganese nodules of the Kara Sea that the enrichment of Fe3+ oxyhydroxides in P is also accompanied by enrichment in HREEs (Vereshchagin et al., 2020). The accumulation of P and REEs in coastal marine sedimentary iron ores is explained by their sorption on Fe3+ oxyhydroxides, which are a result of the coagulation of Fe-bearing colloids from river waters when they are mixed with seawater (Novoselov et al., 2018), and oscillatory fluctuations in redox potential in the coastal marine zone also due to their dilution by river waters (Sokol et al., 2020).
The fine oscillatory zoning of studied rhabdophane aggregates indicates a rapid change in the local growth conditions or the composition of the feeding fluid, which can be due to a rain filtrate percolating from the surface and carrying organic P and desorbed REE (Fink et al., 2016). In the Babaryk occurrence, the periodic fluctuations in pH and compositions of pore solutions after influx of acidic waters and seasonal fluctuations in humidity led to the incorporation of P into the pore water as a result of removal from the soil and desorption from Fe3+. Under favorable conditions, P and REEs form rhabdophane.
Authigenic rhabdophane is found in the shallow-seated brown iron ore nodule of the clayey weathering crust developed at the Babaryk VHMS occurrence, South Urals. Its hexagonal structure is confirmed by EBSD. The mineral forms the fine-grained aggregates, locally, with a radial radiant structure in fractures of colloform Fe3+ oxyhydroxides and spherical aggregates with radial radiant rims and a displaced growth center in a porous goethite matrix. The structure of the rhabdophane aggregates indicates their formation as a result of precipitation from true solutions without recrystallization. Rhabdophane contains LREEs, Ca, S and Y, which is typical of rhabdophane from the laterite weathering crust of granites. The high-quality EBSD patterns of the Ca- and S-rich radial radiant rim of some rhabdophane spherules indicate their isomorphic substitution according to the scheme (Y+REEs)3+ + (PO (Ca+Sr)2+ + (SO4)2−. The rhabdophane-bearing brown iron ore differs from deep-seated brown iron ore of the same borehole in terms of a significant enrichment in LREEs (especially Ce and La) and P. The volcanomictic rocks are believed to be the REE source for the formation of rhabdophane, while P also comes from the soil. Significant enrichment of the brown iron ore in REEs and P and the subsequent formation of relatively large crystalline rhabdophane aggregates are explained by alternating dry and wet periods and processes of REE sorption–desorption. The formation of insoluble REE phosphates with the crystallization of rhabdophane as a result of the surface water circulation and sorption–desorption processes can be one of the stages of the formation of economic REE concentrations similarly to in regolith-hosted REE deposits.
All data concerning measurement and analyses can be found in the article and Supplement. Additional data can be obtained from the corresponding author by request.
The supplement related to this article is available online at: https://doi.org/10.5194/ejm-33-605-2021-supplement.
EVB conceived the work; EVB and KAN undertook sampling; VVS was responsible for EBSD; VVS and IAB handled the SEM and EDA; KAF was responsible for the ICP-MS; EVB was responsible for the XRD; EVB, KAN and VVS were responsible for the writing and original draft preparation.
The authors declare that they have no conflict of interest.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors are grateful to Maksim Lozhkin (Nanophotonics resource center, SPbU) for preparation of the sample surface for EBSD mapping. We also thank Alfons Berger and the two anonymous reviewers for constructive comments on the manuscript.
This research has been supported by the state contract of the SU FSC MG UB RAS no. AAAA-A19-119061790049-3.
This paper was edited by Elena Belluso and reviewed by Alfons Berger and two anonymous referees.
Abedini, A., Asghar Calagari, A., and Masoumi R.: Investigation of mineralogy and geochemistry of kaolin deposit in Kejal area, northwest of Hastjin, Ardebil province, MSc thesis, University of Tabriz, Tabriz, Iran, J. Econ. Geol., 3, 27–35, 2011–2012.
Belogub, E. V.: The mineralogy of alunite-jarosite group in supergene zone of massive suphide deposits of South Urals (Russia), Reunion des Sciences de la Terre 17, Brest: sous l egide de la Societe Geologique de France, 2–4 July, 72 pp., 1998.
Berger, A., Gnos, E., Janots, E., Fernandez, A., and Giese, J.: Formation and composition of rhabdophane, bastnasite and hydrated thorium minerals during alteration: Implications for geochronology and low-temperature processes, Chem. Geol., 254, 238–248, https://doi.org/10.1016/j.chemgeo.2008.03.006, 2008.
Berger, A., Janots, E., Gnos, E., Frei, R., and Bernier, F.: Rare earth element mineralogy and geochemistry in a laterite profile from Madagascar, Appl. Geochem., 41, 218–228, https://doi.org/10.1016/j.apgeochem.2013.12.013, 2014.
Borst, A., Smith, M., Finch, A., Estrade, G., Villanova-de-Benavent, C., Nason, P., Marquis, E., Horsburgh, N., Goodenough, K., Xu, Ch., Kynický, J., and Geraki, K.: Adsorption of rare earth elements in regolith-hosted clay deposits, Nat. Commun., 11, 4386, https://doi.org/10.1038/s41467-020-17801-5, 2020.
Bregiroux, D., Terra, O., Audubert, F., Dacheux, N., Serin, V., Podor, R., and Bernache-Assollant, D.: Solid state synthesis of monazite-type compounds containing tetravalent elements, Inorg. Chem., 46, 10372–10382, 2007.
Clavier, N., Podor, R., and Dacheux, N.: Crystal chemistry of the monazite structure, J. Ceramic Soc., 31, 941–976, 2011.
Dinali, G. S., Root R. A., Amistadi, M. K., Chorover, J., Lopes, G., and Guilherme L. R. G.: Rare earth elements (REY) sorption on soils of contrasting mineralogy and texture, Environ. Int., 128, 279–291, https://doi.org/10.1016/j.envint.2019.04.022, 2019.
Doynikova, O. A., Gorshkov, A. I., Belova, L. N., and Sivtsov, A. V., and Guliy, V. N.: The problem of classification of the phosphates of the rhabdophane group, Zapiski Vseros. Mineral. Obshch., 122, 79–88, 1993 (in Russian with English abstract).
Fink, J. R., Inda, A. V., Tiecher, T., and Barrón, V.: Iron oxides and organic matter on soil phosphorus availability, Ciênc. Agrotec., 40, 369–379, https://doi.org/10.1590/1413-70542016404023016, 2016.
Fu, W., Li, X. T., Feng, Y. Y., Feng, M., Peng, Z., Yu, H. X., and Lin, H. R.: Chemical weathering of S-type granite and formation of Rare Earth Element (REE)-rich regolith in South China: Critical control of lithology, Chem. Geol., 520, 33–51, 2019.
Gallini, S., Jurado, J. R., and Colomer, M. T.: Combustion synthesis of nanometric powders of LaPO4 and Sr-substituted LaPO4, Chem. Mater., 17, 4154–4161, 2005a.
Gallini, S., Jurado, J. R., and Colomer, M. T.: Synthesis and characterization of monazite-type Sr:LaPO4 prepared through coprecipitation, J. Eur. Ceram. Soc., 25, 2003–2007, 2005b.
Hughes, J. C. and le Mare, P. H.: High gradient magnetic separation of some soil clays from Nigeria, Brazil and Colombia, J. Soil Sci., 33, 521–533, 1982.
Hugues, J. M., Foard, E. E., Hubbard, M. A., and Ni, Y.: The crystal structure of cheralite-(Ce), (LREE, Ca, Th, U)(P, Si)O4: a monazite-group mineral, Neues Jb. Miner. Monat., 8, 344–350, 1995.
Ichimura, K., Sanematsu, K., Kon, Y., Takagi, T., and Murakami, T.: REE redistributions during granite weathering: Implications for Ce anomaly as a proxy for paleoredox states, Am. Mineral., 105, 848–859, 2020.
Janots, E., Bernier, F., Brunet, F., Munoz, M., Trcera, N., Berger, A., and Lanson, M.: Ce(III) and Ce(IV) (re)distribution and fractionation in a laterite profile from Madagascar: Insights from in situ XANES spectroscopy at the Ce L-III-edge, Geochim. Cosmochim. Ac., 153, 134–148, 2015.
Kitaev, D. B., Volkov, Y. F., and Orlova, A. I.: Orthophosphates of tetravalent Ce, Th, U, Np, and Pu with the monazite structure, Radiochemistry, 46, 211–217, 2004.
Kozlov, V. I. (Ed.): Geological map of the Russian Federation on a scale of 1: 1000000 (new series) N-40(41), Ufa, 2001.
Krenn, E. and Finger, F.: Formation of monazite and rhabdophane at the expense of allanite during Alpine low temperature retrogression of metapelitic basement rocks from Crete, Greece: Microprobe data and geochronological implications, Lithos, 95, 130–147, https://doi.org/10.1016/j.lithos.2006.07.007, 2007.
Leybourne, M. I., Peter, J. M., Layton-Matthews, D., Volesky, J., and Boyle, D. R.: Mobility and fractionation of rare earth elements during supergene weathering and gossan formation and chemical modification of massive sulfide gossan, Geochim. Cosmochim. Ac., 70, 1097–1112, 2006.
Li, Y. H. M., Zhao, W. W., and Zhou, M. F.: Nature of parent rocks, mineralization styles and ore genesis of regolith hosted REE deposits in South China: An integrated genetic model, J. Asian Earth Sci., 148, 65–95, 2017.
Ling, Q. and Liu, C.: Review of rare earths and fluid–rock interaction, J. Rare Earths, 20, 570–578, 2002.
Makeev, A. B., Borisovskii, S. E., and Krasotkina, A. O.: Chemical composition and age of monazite and kularite from titanium ores of Pizhemskoe and Yarugskoe deposits (Middle and South Timan), Georesources, 22, 22–31, https://doi.org/10.18599/grs.2020.1.22-31, 2020.
Mason, J. and Schuh, C.: Representations of texture, in: Electron Backscatter Diffraction in Materials Science, edited by: Schwartz, A. J., Kumar, M., Adams, B. L., and Field, D. P., Springer, 35–51, 2009.
Mikaeili, K., Asghar Calagari, A., and Abedini, A.: Mineralogical studies in Alibaltalu bauxitic-lateritic deposit of Triassic-Jurassic age, Shahindezh, West-Azarbaidjan, NW Iran. Proceeding of the 17th symposium on Iranian Society of Crystallography and Mineralogy, University of Hamadan, 1–4, 2009.
Moffett, J. W.: Microbially mediated cerium oxidaiton in seawater, Nature, 345, 421–423, 1990.
Montross, S. N., Verba, C. A., Chan, H. L., and Lopano, C.: Advanced characterization of rare earth element minerals in coal utilization byproducts using multimodal image analysis, Int. J. Coal Geol., 195, 362–372, 2018.
Mooney, R. C. L.: Crystal Structures of a Series of Rare Earth Phosphates, J. Chem. Phys., 16, 1003–1003, https://doi.org/10.1063/1.1746668, 1948.
Novoselov, K. A., Belogub, E. V., and Ayupova, N. R.: Babaryk ore field (Alexandrinskoe ore area, South Urals), IMin UB RAS, Miass, Russia, 175 pp., 2009.
Novoselov, K. A., Belogub, E. V., Kotlyarov, V. V., Filippova, K. A., and Sadykov, S. A.: Mineralogical and geochemical features of oolitic ironstones from the Sinara-Techa deposit, Kurgan district, Russia, Geol. Ore Deposit., 60, 265–276, https://doi.org/10.1134/S1075701518030066, 2018.
Novoselov, K. A., Belogub, E. V., Zaykov, V. V., and Yakovleva, V.: Silver sulphotellurides from volcanic-hosted massive sulhide deposits in the Southern Urals, Miner. Petrol., 87, 327–349, https://doi.org/10.1007/s00710-006-0132-x, 2006.
Orlova, A. I., Kitaev, D. B., Volkov, Y. F., Pet'kov, V. I., Kurazhkovskaya, V. S., and Spiridonova, M. L.: Double phosphates of Ce(IV) and some mono and bivalent elements, Radiochemistry, 43, 225–228, 2001.
Orlova, A. I., Kitaev, D. B., Kazantsev, N. G., Samoilov, S. G., Kurazhkovskaya, V. S., and Vopilina, E. N.: Double phosphates of Ce(IV) and some monoand divalent elements: synthesis and crystal structure, Radiochemistry, 4, 326–331, 2002.
Puchkov, V. N.: General features relating to the occurrence of mineral deposits in the Urals: What, where, when and why, Ore Geol. Rev., 85, 4–29, 2017.
Rasmussen, B., Buick, R., Wayne, R., and Taylor, C.: Removal of oceanic REE by authigenic precipitation of phosphatic minerals, Earth Planet. Sc. Lett., 164, 135–149, 1998.
Romero, B., Bruque, S., Aranda, M. A. G., and Iglesias, J. E.: Syntheses, crystal structures, and characterization of bismuth phosphates, Inorg. Chem., 33, 1869–1874, 1994.
Rudmin, M., Reva, I., Sokol, E., Abdullayev, E., Ruban, A., Kudryavtsev, A., Tolkachev, O., and Mazurov, A.: Minerals of rare earth elements in high-phosphorus ooidal ironstones of the Western Siberia and Turgai depression, Minerals, 10, 11, https://doi.org/10.3390/min10010011, 2020.
Scharmová, M. and Scharm, B.: Rhabdophane group minerals in the uranium ore district of northern Bohemia (Czech Republic), J. Czech Geol. Soc., 39, 267–280, 1994.
Scherer, N. C., Engi, M., Gnos, E., Jakob, V., and Liechti, A.: Monazite analysis; from sample preparation to microprobe age dating and REE quantification, Schweiz, Mineral. Petrogr. Mitt., 80, 93–105, 2000.
Seredin, V. V. and Dai, S.: Coal deposits as potential alternative sources for lanthanides and yittrium, Int. J. Coal Geol., 94, 67–93, 2012.
Shelyug, A., Mesbah, A., Szenknect, S., Clavier, N., Dacheux, N., and Navrotsky, A.: Thermodynamics and stability of rhabdophanes, hydrated rare earth phosphates REPO4 •n H2O, Front. Chem., 6, 604, https://doi.org/10.3389/fchem.2018.00604, 2018.
Sigov, A.: Mesozoic and Cenozoic ore formation in Urals “Nedra”, Moscow, Russia, 253 pp., 1969.
Sivakumar, V. and Varadaraju, U. V.: Environmentally benign novel green pigments: Pr1−xCaxPO4(x= 0–0.4), Bull. Mater. Sci., 28, 299–301, 2005.
Škoda, R. and Čopjakova, R.: Authigenic REE minerals from greywackes of the Drahany Uplands, Czech Republic and their significance for diagenetic processes, Mineral. Polonica, 28, 208–218, 2006.
Smith, M. P., Henderson, P., and Campbell, L. S.: Fractionation of the REE during hydrothermal processes: constraints from the Bayan Obo Fe–REE–Nb deposit, Inner Mongolia, China, Geochim. Cosmochim. Ac., 64, 3141–3160, https://doi.org/10.1016/S0016-7037(00)00416-6, 2000.
Sokol, E. V., Kokh, S. N., Kozmenko, O. A., Nekipelova, A. V., Rudmin, M., Khvorov, P. V., and Artemyev, D. A.: Geochemistry and mineralogy of rare earth elements in high-phosphorus ooidal ironstones: A case study of the Kamysh-Burun deposit (Azov–Black Sea iron Province), Ore Geol. Rev., 127, 103827, https://doi.org/10.1016/j.oregeorev.2020.103827, 2020.
Szopa, K., Banasik, K., and Krzykawski, T.: Mechanism of rhabdophane-(La) and lanthanite-(La) formation during reduction of bioavailabe nutrients in water based on SEM and XRD study, Contemp. Trends Geosci., 1, 99–102, https://doi.org/10.2478/ctg-2012-0015, 2012.
Taylor, S. R. and McLennan, S. M.: The continental crust: its composition, and evolution, Blackwell, Oxford, 1985.
Vereshchagin, O. S., Perova, E. N., Brusnitsyn, A. I., Ershova, V. B., Khudoley, A. K., Shilovskikh, V. V., and Molchanova, E. V.: Ferro-manganese nodules from the Kara Sea: Mineralogy, geochemistry and genesis, Ore Geol. Rev., 106, 192–204, https://doi.org/10.1016/j.oregeorev.2019.01.023, 2020.
Yakhontova, L. and Grudev, A.: The mineralogy of oxidized ores: reference book. “Nedra”, Moscow, Russia, 198 pp., 1987.