the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Dec 2024
| 19 Dec 2024
The coesite–stishovite transition of hydrous, Al-bearing SiO2: an in situ synchrotron X-ray study
Monika Koch-Müller
Christian Lathe
Bernd Wunder
Oona Appelt
Shrikant Bhat
Andreas Ebert
Robert Farla
Vladimir Roddatis
Anja Schreiber
Richard Wirth
We examined the influence of Al2O3 and H2O on the position of the coesite–stishovite transition by means of in situ X-ray diffraction measurements with the large-volume press at the PETRA III synchrotron in Hamburg. The position of the transition was determined by several reversal experiments and was found to be shifted almost in parallel by about 1.5 GPa to lower pressures compared to results for the pure SiO2 system reported by Ono et al. (2017). Two further reversal experiments with either additional Al2O3 or additional H2O added to SiO2 showed smaller changes compared to the results of Ono et al. (2017), indicating the effect of the coupled Al and H incorporation in coesite and stishovite on their transition. Further investigations of the solid quenched products and of products from additional multi-anvil experiments performed at the GFZ Helmholtz-Zentrum für Geoforschung in Potsdam were done by powder X-ray diffraction (XRD), transmission electron microscopy (TEM), electron probe micro-analysis (EPMA), and Fourier transform infrared (FTIR) and Raman spectroscopy. Generally, the recovered samples of the in situ experiments contained less stishovite than expected from the last in situ XRD pattern before quenching. Thus, these investigations clearly show that hydrous, Al-rich stishovite that formed at high pressure (P) and temperature (T) could, at least partly, not be quenched to room conditions and transformed to coesite with unusually high (Al, H) contents. As result of this, conventional quench experiments would lead to erroneous results of the transition in the (Al, H)-bearing SiO2 system. We observed two kinds of coesite in the experiments: one relatively Al-poor coesite transformed under equilibrium conditions at P and T from stishovite over a certain time frame and an Al-richer one, sometimes pseudomorphically replacing former stishovite during the decompression process to room conditions. Within both types of coesite, nanometre-sized kyanite inclusions and relicts or remnants of stishovite were observed by TEM. These observations resemble those of Yang et al. (2007) on ophiolites with identical textures and phases and were interpreted as result of a stishovite transition back to coesite during retrograde metamorphism. Our results clearly indicate that the coesite–stishovite transition is sharp but can considerably vary in depth by the addition of Al and H to the SiO2 system. This has consequences for the assignment and interpretation of the depth variation of the seismic X discontinuity.
- Article
(2918 KB) - Full-text XML
-
Supplement
(7581 KB) - BibTeX
- EndNote





For more than 50 years, the pressure–temperature (P–T) conditions of the transition of the two silica polymorphs coesite (Coe) and stishovite (Sti) have been investigated in numerous experimental studies in the pure SiO2 system (e.g. Akimoto, 1972; Yagi and Akimoto, 1976; Zhang et al., 1996; Akaogi et al., 2011; Ono et al., 2017). Using a multi-anvil apparatus in combination with in situ synchrotron radiation, Ono et al. (2017) determined the Coe–Sti equilibrium to be P (GPa) = 4.7 + 0.0031×T (K). Nowadays, this equation is used in many multi-anvil labs for pressure calibration at high temperatures; thus, it seems to be accepted by the community to be the most accurate one. The Coe–Sti transition is not only important for reasons of lab calibration but also controversially discussed as a cause of the seismic X discontinuity (e.g. Williams and Revenaugh, 2005; Knapp et al., 2015; Srinu et al., 2021; Pugh et al., 2021). The X discontinuity is not a global feature and has been observed in several regions of active mantle dynamics, such as subduction zones or beneath oceanic plates within the upper mantle in the depth range of 230–350 km (Revenaugh and Jordan, 1991). The Coe–Sti transition is associated with a dramatic change in elastic properties (Chen et al., 2015) and a density jump of about 50 % from Coe having a structure with silicon in tetrahedral coordination ([4]Si) to Sti having octahedral Si coordination ([6]Si, rutile structure). Thus, the shear and compressional wave velocities in Sti exceed those of Coe by more than 30 % (Weidner et al., 1982), and as a consequence, only a few volume percentage points of free SiO2, e.g. occurring in deeply subducted oceanic (eclogitic) crust, would be needed to cause a visible seismic discontinuity from this transition (Williams and Revenaugh, 2005). At conditions of the Coe–Sti boundary, it is assumed that the abundance of SiO2 in subducted oceanic and continental crust is about 10 and 20 vol. %, respectively (Litasov et al., 2007). However, results of a recent experimental study (Knapp et al., 2015) question the existence of free SiO2 in deep eclogites and thus challenge the SiO2 transition as plausible explanation of this seismic discontinuity.
It is assumed that the Coe–Sti transition is sharp because SiO2 is thought to be a near-pure component in recycled basalt (Zhang et al., 1996). However, the observed depth variation of the X discontinuity cannot be explained with such a sharp single mineralogical change, as this would require too large a spatial variation in the mantle temperature. Therefore, to explain the depth range of more than 100 km observed for the X discontinuity, possible candidates, beside the SiO2 transition, have been discussed for explaining these seismic reflectors: the formation of phase A (Liu, 1987); transformation from orthoenstatite to high-P clinoenstatite (Angel et al., 1992; Woodland, 1998); reaction of forsterite + periclase to anhydrous phase B (Ganguly and Frost, 2006); occurrence of carbonated silicate melt (Dasgupta et al., 2013); and formation of the 3.65 Å phase (Mookherjee et al., 2015). However, as emphasized by Pugh et al. (2021) and Srinu et al. (2021) none of these candidates alone fulfils the requirements of the seismic observations; thus they suggest multiple causes for explaining the X discontinuity.
Coesite and stishovite can incorporate significant amounts of Al and H in their structures by various substitution mechanisms. For Coe the “hydrogarnet” substitution ([4]Sivacancy + 4H+) is the most relevant, with the coupled substitution ([4]SiAl H+) being relevant to a minor extent (e.g. Koch-Müller et al., 2001, 2003); for Sti more mechanisms are described including coupled substitution at the octahedral position ([6]SiAl H+), Al incorporation via oxygen vacancies (2 [6]Si [6]Alvacancy), hydrogarnet substitution at the octahedral position ([6]Sivacancy + 4H+), incorporation of interstitial molecular water (SiO2 × H2O), and Al incorporation at interstitial octahedral sites (e.g. Pawley et al., 1993; Bromiley et al., 2006; Litasov et al., 2007; Spector et al., 2016). The maximum amounts of H and Al that can be incorporated in Sti significantly exceed that of Coe. For Coe the maximum storage capacities of H and Al were found to be 200 ppm (expressed as wt % H2O) and 17 wt % ppm Al (Koch-Müller et al., 2001, 2003); for Sti maximum amounts were determined to be 32 000 wt % ppm H2O (Nisr et al., 2017) and 7.6 wt % Al2O3 (Litasov et al., 2007). More complexity is accomplished as the amounts of Al and H incorporation strongly vary with P and T (e.g. Litasov et al., 2007; Purevjav et al., 2024; Chen et al., 2024) and as initially formed hydrous Sti is metastable and dehydrates over time (Spector et al., 2016). According to Kueter et al. (2023) about half of the water incorporated in Sti at high P and T cannot be quenched experimentally to room conditions.
At conditions of the lower mantle, stishovite transforms to post-stishovite with a CaCl2 structure (e.g. Lakshtanov et al., 2005, 2007). Post-stishovite is able to incorporate Al and H into their structure to amounts exceeding that of Sti (e.g. Litasov and Ohtani, 2005; Litasov et al., 2007). The incorporation of Al and H into these SiO2 polymorphs modifies their phase diagrams in that the pressure of the rutile–CaCl2-type phase transition is shifted to lower pressure (e.g. Lakshtanov et al., 2005, 2007). More precisely, the transition pressure is reduced from about 80 GPa to less than 30 GPa at 2500 K by the incorporation of 3 wt % Al2O3 into Sti (Bolfan-Casanova et al., 2009). By analogy, recent reports show that phase stabilities of nominally Al-free hydrous Mg silicates like antigorite (Bromiley and Pawley, 2003) and superhydrous phase B (Li et al., 2022a, b) and phase D (Pamato et al., 2015; Xu et al., 2021) are also significantly changed by additional incorporation of Al (and H).
Surprisingly, the influence of (Al, H) incorporation into Coe and Sti on their transition has, to our knowledge, not been investigated, yet; thus, the main intention of this study was to get more insight into this by applying an experimental approach.
2.1 High-pressure equilibrium experiments
We performed seven in situ and three quench high-pressure multi-anvil experiments (Tables 1, 2) at the synchrotron storage ring, at the Deutsches Elektronen-Synchrotron (DESY), Hamburg, and the GFZ Helmholtz-Zentrum für Geoforschung, Potsdam, respectively. Starting materials were included for all 10 high-pressure experiments, but 2 consisted of a mixture of amorphous SiO2 plus 1 wt % γ-Al2O3 plus 10 wt % H2O (mix 1). For one equilibrium experiment we used only amorphous SiO2 plus 1 wt % γ-Al2O3 without water (mix 2, BT-785), and one was performed with SiO2 plus water only (mix 3, BT-786).
Table 1Results of in situ high-pressure experiments at DESY, including their starting material, experimental conditions, and reaction progress. Ia is the intensity of the diffraction peaks when the target temperature was reached, and I0 is the intensity of the diffraction peaks from the previous measurement.
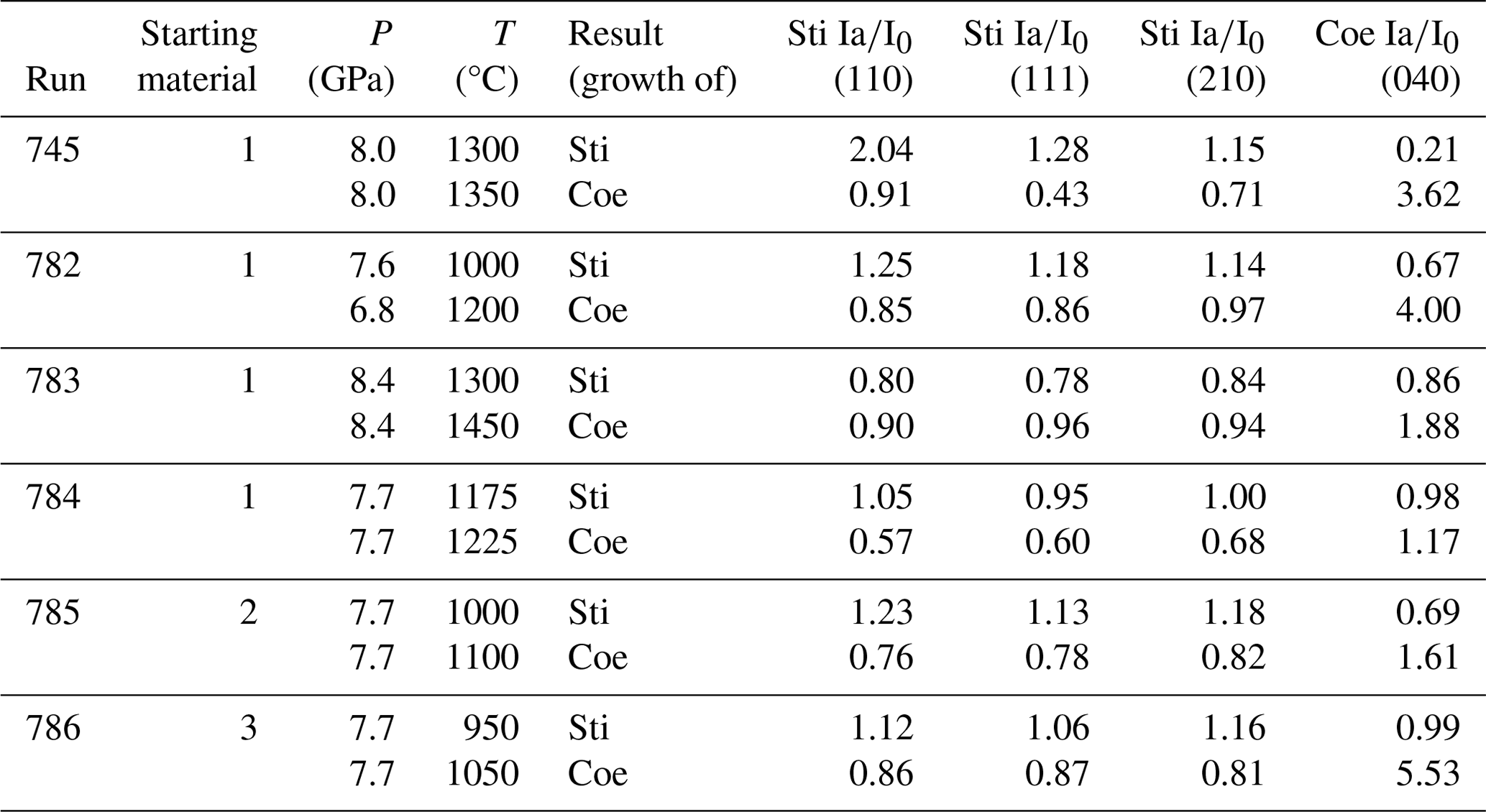
l Mix 1: 99 wt % SiO2, 1 wt % Al2O3 (both amorphous), 10 wt % H2O. Mix 2: 99 wt % SiO2, 1 wt % Al2O3 (both amorphous). Mix 3: SiO2 (amorphous), 10 wt % H2O.
Table 2Experimental details, results, and analysis methods used for samples of the conventional quench experiments at GFZ.
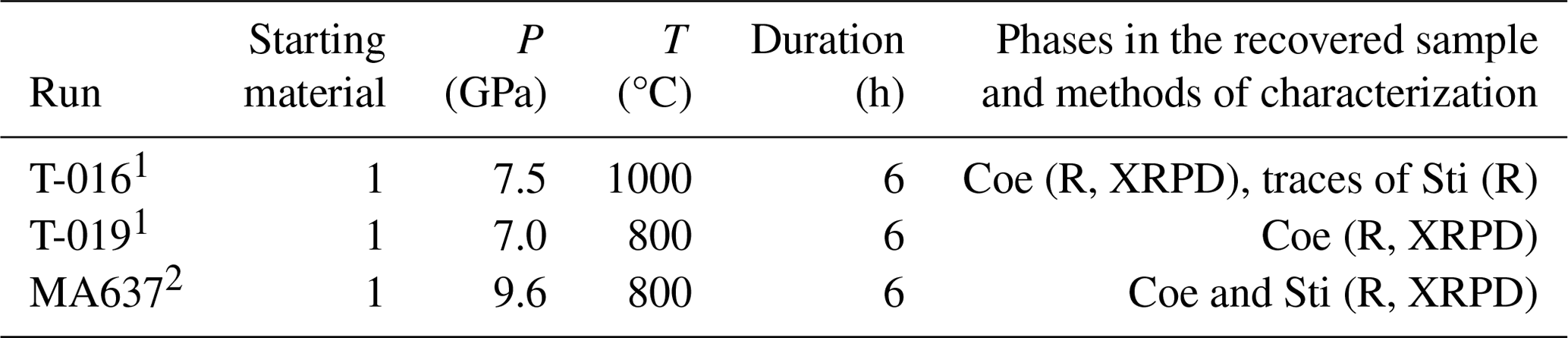
1 Combined piston-cylinder–multi-anvil press at GFZ. 2 Rotating multi-anvil press at GFZ. R: Raman spectroscopy; XRPD: X-ray powder diffraction. For the starting material of the experiments, see Table 1.
In all runs we used Ti capsules (2 mm long and 1.4 mm inner diameter) which are X-ray transparent and can be sealed watertight (Chinnery et al., 1999). However, after the experiments the material was very brittle and it was not possible to verify by direct methods that the water was kept inside the capsule during the experiment. Therefore, the presence of water was indirectly verified by collecting Fourier transform infrared (FTIR) spectra of the recovered phases in the OH-stretching region: in all but one experiment, using mix 1 and mix 3, the recovered phases did show incorporation of water. The one experiment (with mix 1 as the starting material), which was considered to be dry, was deleted from the list of experiments (Table 1) as it also showed deviant behaviour in respect to chemistry (much less Al incorporation) and in respect to the phase stability (no shift observed). All but one of the recovered Ti capsules (or parts of it) with parts of the very compact sample attached were embedded in epoxy, ground to expose the surface of the sample, and polished. In this kind of preparation, the original texture is still preserved and can help to figure out the ongoing processes. In one experiment (BT-783) several relatively large (100–200 µm) hand-picked compacted grains of the sample were embedded in epoxy and polished. Thus, the original texture was not fully preserved here. All samples were investigated by Raman spectroscopy for phase identification; electron probe micro-analysis (EPMA) and backscattered-electron (BSE) images (point analyses of Al, Si, Ti; Al, Ti element mapping) for composition and textural observations; and Fourier transform infrared (FTIR) spectroscopy for H incorporation and quantification. Some samples were investigated by X-ray powder diffraction (XRPD) and transmission electron microscopy (TEM) for structural and chemical information (high-resolution episcopic microscopy, HREM; energy-dispersive spectroscopy, EDS; electron energy loss spectroscopy, EELS).
2.1.1 In situ multi-anvil press experiments
The six successful in situ multi-anvil experiments were carried out at beamline station P61B, PETRA III, at DESY in Hamburg, Germany. The synchrotron was operating in a top-up mode with an energy of 6 GeV and current of 120 mA. The high-energy white-beam beamline was operated using energy-dispersive (ED) X-ray diffraction (XRD) with Ge solid-state detector (SSD) and a multichannel analyser (MCA) to collect the diffraction patterns in the energy range of 30–160 keV. The channel-to-energy relationship of the SSD MCA was calibrated using the gamma ray lines of different metals (57Co and 133Ba). The large-volume press, a Hall-type six-ram one (mavo press LPQ6 1500-100; Max Voggenreiter GmbH, Germany), can achieve a maximum combined press load of 1500 t (∼ 620 bar oil pressure in each of the six hydraulic rams).
The design of the experimental high-pressure assembly was the same as we use at GFZ. It consisted of a MgO octahedron with a cylindrical drilling and an edge length of 14 mm. We used an octahedron made of MgO and doped with 5 wt % Cr2O3 to lower the thermal conductivity at high temperature. The drilling hole contained a stepped graphite heater, pressure-transmitting MgO parts, the sample container, and the type C thermocouple (TC). The pressure calibration and temperature distribution within this assembly are discussed in the next subsection. The octahedron was then surrounded by eight tungsten carbide cubes with truncations of 8 mm on the edges which can be arranged in a way that the cubes build an octahedral vacancy where the octahedron fits in very well. With that, we built a big cube (made up by eight smaller cubes; called 14/8 assembly), which was placed into the press and was pressurized by remote control from outside the experimental hutch by putting a load on the six hydraulic ramps.
The diffraction angles at which the detectors were positioned were calibrated using the LaB6 660c NIST standard. The pressure was measured directly in situ using MgO as the pressure-transmitting material located just outside the sample container. We used the calibration Fit3-3BM equation-of-state (EOS) model reported by Tange et al. (2009) to calculate the pressure with a precision of ±0.1 GPa, measuring multiple times under similar conditions. The detector positioned at a 3° diffraction angle was used to analyse the 111, 200, and 220 reflections of periclase by fitting the peak positions at room pressure and at each pressure and temperature change with the symmetric pseudo-Voigt function using the open-source software Fityk (Wojdyr, 2010), implemented in the programme PDIndexer ver4.441. The PDIndexer was used to visualize and analyse all the diffraction patterns. The error in pressure calculation is up to ±0.05 GPa depending on the P–T conditions compared to for example Dewaele et al. (2000). We assume that the thermocouple reports the sample temperature accurately; hence the error in the pressure from minor temperature variations (1–2 K) should be small. There is no reason to assume a large discrepancy between the calibrated pressures using press load experiments, such as the Coe–Sti transition as done in conventional multi-anvil experiments and the EOS methods as performed at synchrotron facilities. This is supported by the results of multiple phase stability studies determined in situ compared to literature data (e.g. Lathe et al., 2022). In addition, as a side effect, the Ti capsules can be used as an additional pressure sensor: we observe in the pressure range of 9.0–9.8 GPa hexagonal ω Ti at room T and in the range of 8.0–10.0 GPa and 700–900 °C the cubic β Ti modification, which is perfectly in line with the report by Dewaele et al. (2015).
Pressure measurements were taken multiple times as we adjusted temperature and pressure to reach our target conditions. We also monitored pressure during the annealing processes and noted slight decreases in pressure with increasing temperature and calculated a pressure value for each (P,T) step (see Table S1, Figs. 1 and 2).

Figure 1P–T diagram showing our experimental results (for run numbers, see Table 1) of the in situ experiments. Closed squares (stars) indicate growth of Coe during T increase, while the open symbols indicate re-growth of Sti after Coe growth and T decrease. The half-filled symbols indicate no reaction (Tables 1 and S1). For comparison the results of the experimental study of Ono et al. (2017) with a pure SiO2 system are shown. The black line through the graph labelled “this study” is the new boundary between Sti and Coe in the (H, Al)-bearing SiO2 system. The Coe–Sti transition in the (Al, H)-bearing SiO2 system is shifted by about 1.5 GPa at lower pressures to be almost parallel to Ono et al. (2017). Experiments with adding water (no Al2O3) to SiO2 (BT-786) and with adding Al2O3 (no water) to SiO2 (BT-785) show shifts of about 0.8 GPa at lower P compared to Ono et al. (2017).
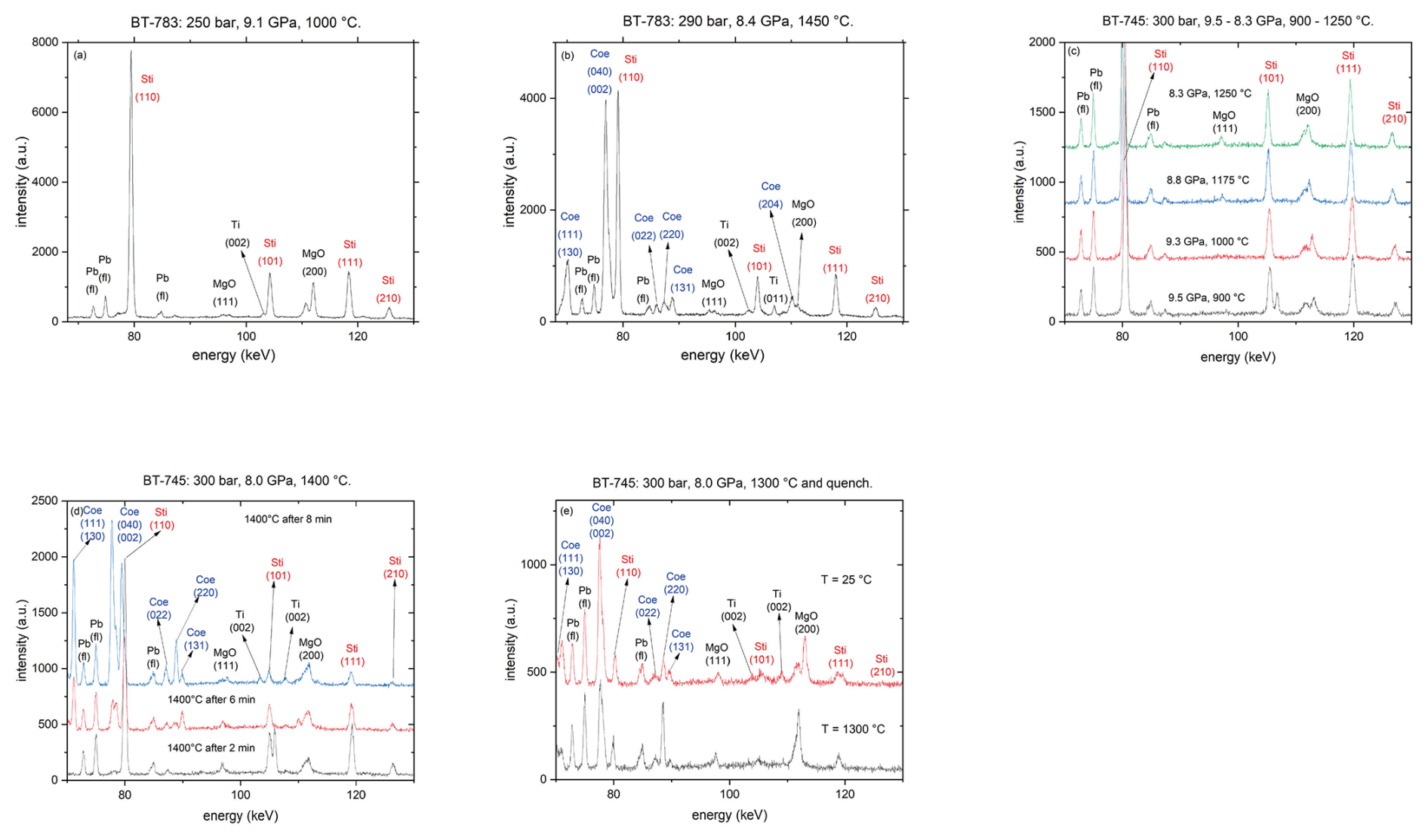
Figure 2Synchrotron energy-dispersive XRD pattern (diffraction angle of 3°; a.u. denotes arbitrary units) (a, b) run BT-783 after Sti synthesis at 9.1 GPa and 1000 °C and shortly before T quenching at 8.4 GPa and 1450 °C, respectively; (c) run BT-745 from 900–1250 °C; (d) run BT-745 at 1400 °C as a function of time; and (e) run BT-745 before quenching at 1300 °C and after quenching to room T.
Our experimental approach is outlined in Fig. S1 in the Supplement: we compressed the assembly at room T to a target pressure between 8 and 10 GPa (path 1), increased temperature to 800–1000 °C (path 2) and remained there for about 30 min to synthesize Sti (point 3), and after a successful Sti synthesis further increased the temperature (path 4) until we observed the first indication for Coe growth (appearance of the (040) and (002) reflections). To do so, we held the temperature at a certain pressure for some time while measuring XRD patterns of the sample to determine the phase relations (growth of Coe). As pressure drops during heating, we also took XRD patterns of the pressure sensor for MgO to determine the pressure by analysing the P-induced shifts in MgO reflections. As soon as we observed the first indication of Coe growth we bracketed the Coe-to-Sti transformation reaction at the given P by decreasing temperature to monitor the growth of Sti (appearance of the 110 reflection) and breakdown of Coe. The criteria for Coe growth were based on the calculation of the transformed volume of Coe to Sti by analysing the relative peak intensities of the overlapping diffraction peaks (040), (20-2), and (002) of Coe. Additionally, we used the reflections 110, 111, and 210 of Sti, which resulted in similar growth rates. The degree of transformation was calculated from the ratio of , where Ia is the intensity of the diffraction peaks when the target temperature was reached and I0 is the intensity of the diffraction peaks from the previous measurement (see Table 1). The intensity of the peaks was determined by fitting the diffraction pattern with the symmetric pseudo-Voigt function in the open-source programme PDIndexer ver4.441. The whole procedure from the Sti synthesis until the final quenching could take up to 4 h (see Table S1), while the sample was under very slowly increasing temperature and decreasing pressure. Finally, we quenched the experiment by shutting off the heating system and decreasing the temperature (path 5) to T of less than 100 °C within seconds and released pressure over at least 8 h (path 6).
Importantly, we observed that in some experiments (Table S1) small amounts of Coe crystallized during early stages of heating at a given pressure high enough to be in the stability field of Sti. This is in line with the observations of Yagi and Akimoto (1976), who observed formation of metastable Coe at approximately 200 °C, when starting the experiments with amorphous SiO2, which is also the case in our experiments. We observed that the metastably formed Coe disappeared again, when keeping the experiment at higher T within the stability field of Sti for some time period.
2.1.2 High-pressure multi-anvil quench experiments
Three high-pressure experiments were performed using the multi-anvil presses at GFZ Potsdam. To be consistent with the in situ experiments performed at beamline P61B, we used the same assembly (including the Ti capsules) as described above. The pressure assembly was calibrated at GFZ with the rotating multi-anvil press against the phase transitions in Bi metal (Lloyd, 1971; Piermarini and Block, 1975). Calibrations at higher temperature are based on the following phase transitions: CaGeO3 garnet–perovskite (Susaki et al., 1985), SiO2 coesite–stishovite (Ono et al., 2017), and Mg2SiO4α–β (Morishima et al., 1994). Estimated pressure uncertainty is about ±0.3 GPa. The precision of the temperature measured by the TC is ±10 °C. As this type of TC can easily undergo oxidation, we surrounded the tip of the TC with BN powder to prevent oxidation. The absence of a temperature gradient was verified by applying the 2-pyroxene geothermometer (see Müller et al., 2017). Within the 2 mm long capsule the temperature was homogenous distributed at ±30 °C. The variation is not due to a real temperature variation but due to the limitation of the electron microprobe (EMP) analyses (mixed analyses).
2.2 Analytic methods
2.2.1 X-ray powder diffraction analysis
Some of the quenched samples were ground; diluted with Elmer's white glue; and evenly spread on a circular foil, which was then covered with a second empty foil. X-ray powder diffraction patterns of these samples were recorded in transmission with a fully automated STOE STADI P diffractometer using CuKα1 radiation at 40 kV and 40 mA (take-off angle of 6°), with a primary monochromator and a 7° wide position-sensitive detector. The intensities were recorded between 5 and 125° 2θ with a detector step size of 0.05° and a resolution of 0.02°. The collected patterns were processed using the GSAS software package for Rietveld refinement (Larson and von Dreele, 2004) for phase identification, phase proportions, and unit-cell parameters. Initial crystal structures were from Geisinger et al. (1987) for Coe and from Kroll and Milko (2003) for Sti, both taken from the Inorganic Crystal Structure Database (ICSD; FIZ Karlsruhe, https://icsd.fiz-karlsruhe.de/, last access: 17 December 2024). Results are summarized in Table S2.
2.2.2 EPM (electron microprobe) analyses
Electron probe micro-analysis (EPMA) using wavelength and/or energy-dispersive spectroscopy (WDS and EDS) and backscattered-electron (BSE) images was performed with the JEOL Superprobe (JXA-8230). We performed Si and Al point analyses on the cross sections of the recovered capsules on all nine in and ex situ experiments and Al and Ti mapping and energy-dispersive spectroscopy for Ti on two in situ experiments: BT-745 and BT-784. The analytical conditions included an acceleration voltage of 15 kV, a beam current of 10 nA, and a beam diameter of 1 µm. As is standard for Si, Al, and Ti, we used well-defined synthetic Coe, orthoclase, and rutile. On-peak and background counting times for Si were 10 and 5 s, respectively, and 20 and 10 s for Al and Ti.
2.2.3 Raman spectroscopy
Raman spectra of different solid quenched phases were collected with a HORIBA Jobin Yvon LabRAM HR800 VIS spectrometer (GFZ, Potsdam) equipped with a green 514 nm wavelength diode-pumped solid-state laser. The spectra were taken from the microprobe mounts for phase identification after EMPA and subsequent removal of the carbon coating. Spectra were measured in the range of 100–1000 cm−1, with a data acquisition time of 10–20 s, with three accumulations and 50 % laser power. The spectra were deconvoluted with the software PeakFit by Jandel Scientific. Unfortunately, spectra in the OH-stretching region could not be measured by Raman spectroscopy due to high fluorescence of the samples and sensitivity of the samples to the laser beam.
2.2.4 FTIR spectroscopy
FTIR spectra were measured using a VERTEX 80v FTIR spectrometer (Bruker Optics) with an attached Hyperion II microscope at the IR spectroscopy laboratory of the GFZ in Potsdam. A tungsten light source, InSb detector, and CaF2 beam splitter were used to measure the OH-stretching range (2500–4000 cm−1). We collected unpolarized single-crystal spectra on the randomly oriented Coe and Sti formed at relatively low temperature (800 °C) with thicknesses of about 20 to 50 µm. Due to the higher water content of the Coe and Sti equilibrated at high temperature (e.g. 1450 °C), for these samples we applied the thin-film method to determine the absorption pattern (e.g. Li et al., 2022). The spectra were deconvoluted with the software PeakFit by Jandel Scientific. We applied a Gaussian plus Lorentzian peak shape to all component bands. The integral absorbance (Aint) of each peak was summed up and multiplied by 3 to get the total integral absorbance Aint,tot. We used the Beer–Lambert law and the calibrations by Thomas et al. (2009) and Koch-Müller et al. (2001) to calculate the hydrogen content expressed as wt % ppm H2O for Coe and Sti, respectively.
2.2.5 TEM analyses
Cross-sectional specimens (TEM foils) for TEM investigations were prepared by a lift-out method with a Thermo Fisher Scientific Helios G4 UC focused ion beam (FIB) (Ga) device operated at 30, 16, 5, and 2 keV. TEM and scanning TEM (STEM) investigations were conducted with an FEI Tecnai F20 microscope operating at an accelerating voltage of 200 kV. In addition to imaging selected-area electron diffraction (SAED) patterns were collected to verify various SiO2 phases. Electron energy loss spectroscopy (EELS) and energy-dispersive X-ray (EDX) spectroscopy were conducted on a probe Cs-corrected Themis Z 3.1 60–300 kV (Thermo Fisher Scientific) microscope operated at 300 kV and equipped with a Super-X detector EDX system and a Gatan Continuum 1065 ER spectrometer. EEL spectrum images were recorded with 1.5 and 0.15 eV per channel dispersion using a dual-EELS mode selecting a valence EELS and oxygen K-edge energy windows.
3.1 Phase relations
Based mainly on four of the in situ experiments using mix 1, we constructed a new phase diagram (Fig. 1), showed our brackets, and deduced a Coe-to-Sti transformation line in comparison to the experimental study of Ono et al. (2017) for the pure SiO2 system. We have chosen Ono et al. (2017) as the reference for the pure SiO2 system as this study is widely accepted by the community to be the most accurate one. In addition, it served among others as a calibrant for the multi-anvil presses at GFZ. The black line labelled “this study” is a simplification of the new boundary between Coe and Sti in the (Al, H)-bearing SiO2 system, as the data do not allow for distinguishing between a straight or curved line (see below in the Discussion). The Coe-to-Sti transition in the (Al, H)-bearing SiO2 system is shifted almost parallel to Ono et al. (2017) by about 1.5 GPa for lower pressures. Two further reversal experiments, one with only additional H2O added to SiO2 and a second one with additional Al2O3 and no water, were performed. They both are shifted by about 0.8 GPa from the findings of the pure system by Ono et al. (2017) to lower pressures in the direction of the boundary found in this study. These observations indicate that it is the combination of Al2O3 and water that seems to be relevant for causing the observed P–T shift of the transition. For experiments performed at the lowest pressures and temperatures (BT-782) the transition from Coe to Sti was kinetically hampered, even using excess water in the capsule. Thus, over a wide (P,T) range no reaction was observed (half-filled symbols in Fig. 1). This is in line with the observations made by Zhang et al. (1996), performing experiments in a dry, Al-free system and observing extreme sluggishness of the Coe–Sti reaction at low T; thus no bracketing was possible at T below 1000 °C in their study.
3.1.1 Details of the in situ experiments at DESY
Figure 2a shows a representative diffraction pattern for the Sti synthesis of run BT-783 at 9.1 GPa and 1000 °C. Beside the Sti reflections, other contributions from the Ti capsule, MgO parts, and Pb fluorescence lines from the detector are visible. Figure 2b shows the same experiment 168 min after the synthesis shown in Fig. 2a and shortly before quenching at 8.4 GPa and 1450 °C with coexisting Coe and Sti. Figure 2c shows a series of diffraction patterns of run BT-745 with increasing temperature and decreasing pressure: the diffractograms taken at 9.5 GPa and 900 °C, 9.3 GPa and 1000 °C, 8.8 GPa and 1175 °C, and 8.3 GPa and 1250 °C show only Sti even after holding P and T for 11, 19, 8, and 14 min, respectively. At 8 GPa and 1400 °C the first occurrence of Coe can be observed after 2 min (Fig. 2d), which becomes more clear with time – after 8 min at 8 GPa and 1400 °C the amount of Coe exceeds that of Sti (Fig. 2d). Thus, at these conditions the transformation reaction from Coe to Sti in the presence of water and Al2O3 is extremely fast. After observation of the growth of Coe we aimed for bracketing the Coe–Sti transformation at this P by decreasing T stepwise and monitoring the reaction. Figure 2e shows the final in situ diffraction patterns at 8 GPa and 1300 °C shortly before T quenching together with the one after T quenching to room T (estimated P of ca. 5 GPa). The weight percent (wt %) ratio of Sti:Coe was about 1:4. This was our first experiment, and to facilitate the analyses of the recovered phases we adjusted for runs BT-782 to BT-786 the P–T conditions after bracketing the phase transition so that the Coe–Sti ratio of the quenched samples was about 1:1, as shown in Fig. 2b for run BT-783.
Experimental conditions and results of all in situ experiments are shown in Tables 1 and S1.
3.1.2 Conventional quench experiments
The experimental details and results for the three conventional quench experiments are given in Table 2. All three experiments were performed with mix 1 in the SiO2–Al2O3–H2O system. In contrast to the in situ experiments the quench experiments were conducted at relatively low temperature and thus annealed more or less isobarically to the target temperature and held at the specific P–T conditions for 6 h.
3.2 Characterization of the recovered run products
Characterization of the quenched solids was aimed to determine texture; phase relations of formed phases; (Al, H) contents of the SiO2 phases; potential Ti contaminations from the capsule material; and phase portions and properties, e.g. cell dimension of coesite and stishovite. Generally, the recovered samples of the in situ experiments contained less Sti than expected from the last in situ XRD pattern before quenching. This observation is mainly based on Raman spectroscopy of all experiments (Raman mapping of the sample mounts in the range of 450–800 cm−1 with more than 100 spectra) and XRPD of sample BT-783 (Table S1) compared to in situ diffraction.
3.2.1 Texture and phase relations
Figure 3a shows a BSE image of sample mount BT-745 with part of the Ti capsule (light), grey-coloured grains of the sample, and an Al mapping from EPMA included in the BSE image at the position where it was taken. In the BSE image we observed no visible difference between the grains, and with Raman spectroscopy (more than 100 spectra) we determined only Coe, with no indication for Sti. However, if we recall the history of this experiment, the Sti:Coe ratio shortly before quenching was 1:4; thus, it is hard to believe that we do not see any Sti. In the Al mapping we distinguished between Al-poor Coe (blue colour) and Al-richer Coe (green colour) together with some red spikes, representing another Al- and Si-rich phase. We have cut two FIB foils for TEM investigations and confirmed with HREM and EDS the presence of two kinds of Coe with different Al contents, and our working hypothesis was that the relatively Al-poor Coe transformed under equilibrium conditions over a certain time period from Sti and that the relatively Al-richer Coe represents former Sti, which transformed to Coe during the abrupt ending of the experiment – thus Sti could not be quenched to room conditions. Within both types of coesite, representing former Sti, nano-sized kyanite (Ky) inclusions were observed by transmission microscopy (red spikes in the Al mapping). The TEM results are documented in Fig. S2a to h in the Supplement. The inclusions are visible in the backscattered-electron (BSE) images and the bright-field (BF) images as round or elongated nanometre- to micrometre-sized particles of light and dark colours, respectively (Fig. S2a–c). Using the combination of energy-dispersive spectroscopy (EDS) and electron energy loss spectroscopy (EELS) mapping (see the section on EELS in the Supplement), it turned out that, beside Ky, some of these inclusions are Sti surrounded by a shell of amorphous SiO2 (Fig. S2d–f). In Fig. S2e and f we show the locations and results of the energy-dispersive analyses of Ky, Sti, and Al-bearing and Al-free Coe; the phase identification was based not only on the results of the energy-dispersive spectra and HREM but also on the results of EELS mapping for Ky, Sti, and Coe (see the section on EELS in the Supplement). Representative electron diffraction patterns for Al-bearing Coe and Ky are shown in Fig. S2g and h, respectively.
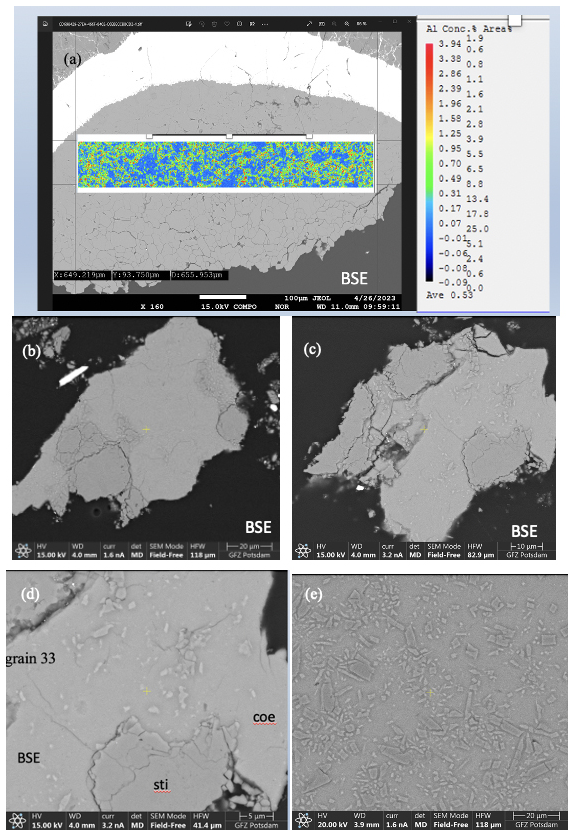
Figure 3(a) BSE image of sample mount BT-745 together with Al mapping. (b–d) BSE images of two different grains from sample BT-783 embedded in epoxy and polished. (e) BSE image of a part of sample mount BT-784. Note in all four BSE images the slightly lighter matrix, consisting of Coe, and the darker grains, consisting of Sti, as revealed by Raman spectroscopy. Panels (d) and (e) show that the Coe matrix is full of smaller inclusions/crystals.
Figure 3b to e show BSE images of the sample mounts of BT-783 and BT-784 at different magnifications. Figure 3b and c show two of the hand-picked, embedded, and polished compact grains of sample BT-783, with the later presented under higher magnification in Fig. 3d. We can distinguish larger darker grains inside a kind of a matrix of a lighter-grey colour. With Raman spectroscopy we identified the matrix as Coe and the darker grains/crystals as Sti. Sti appears darker in the BSE images than Coe (Z contrast – see Discussion), and in images taken with reflected light the opposite holds true (higher refractive index of Sti compared to Coe; 1.8 versus 1.6 for Sti versus Coe). Thus, applying both imaging tools the distribution of Coe and Sti, if both are large enough, can easily be determined. Figure 3e shows sample mount BT-784 (it is similar to that of BT-782; not shown): both BT-782 and BT-784 consist of a lighter-grey matrix of Coe (as revealed by Raman spectroscopy), however with crystals/fragments of only slightly different shades of grey in it. The Raman spectra of the crystals often showed contributions from Coe and Sti (Fig. 4) and appear to be lighter (darker) in the BSE (reflected light) images, respectively. Judging from the colours in Fig. 3d in the Coe matrix we distinguish additional nanometre- to micrometre-sized lighter and darker inclusions. From the sample mounts of BT-783 and BT-784 we cut FIB foils for further TEM investigations such as HREM, EDS, and EELS (see also the EELS section in the Supplement and Fig. S4). TEM investigations of the FIB foils revealed the existence of Coe, Sti, and Ky in both samples. The electron diffraction pattern of the selected larger crystals of sample BT-783 and BT-784 can without a doubt be indexed as Sti. The representative electron diffraction patterns of sample BT-784 are shown in Fig. S3. We applied EELS spectroscopy – more specifically EELS mapping – to investigate the phase and size distribution of Coe, Sti, and Ky in the FIB foils of these two samples – this is explained in detail in the Supplement (Fig. S4). Figure 5 shows parts of the foils for BT-784 (no. 7435) and BT-783 (no. 7479), respectively. FIB foil no. 7435 was cut through the matrix and crystals of various sizes. FIB foil no. 7479 was cut in the Coe matrix through the light and darker inclusions, which continue as expected into the depth and show a worm-like shape in addition to some round inclusions. It turned out that in both samples the matrix is Coe as expected from the Raman spectra, and the nanometre- to micrometre-sized crystals as well as the worm-like inclusions are Sti. If we recall the history of this sample, at certain P and T it consisted of 100 % Sti and after bracketing the Coe–Sti transformation reaction the Sti:Coe ratio was 1:1. The nanometre-sized Sti of both samples shown in Fig. 5 looks like remnants or relicts of former larger Sti crystals, which could only partially be quenched to room conditions but otherwise transformed to Coe. In all samples the Coe matrix is full of Ky inclusions which look like exsolutions from a former Al-richer Sti. This is illustrated in Fig. 5 in the Al mapping for sample BT-784 and in the EELS mapping for sample BT-783. In addition, the Al mapping in Fig. 5 shows Al enrichment in Sti compared to Coe.
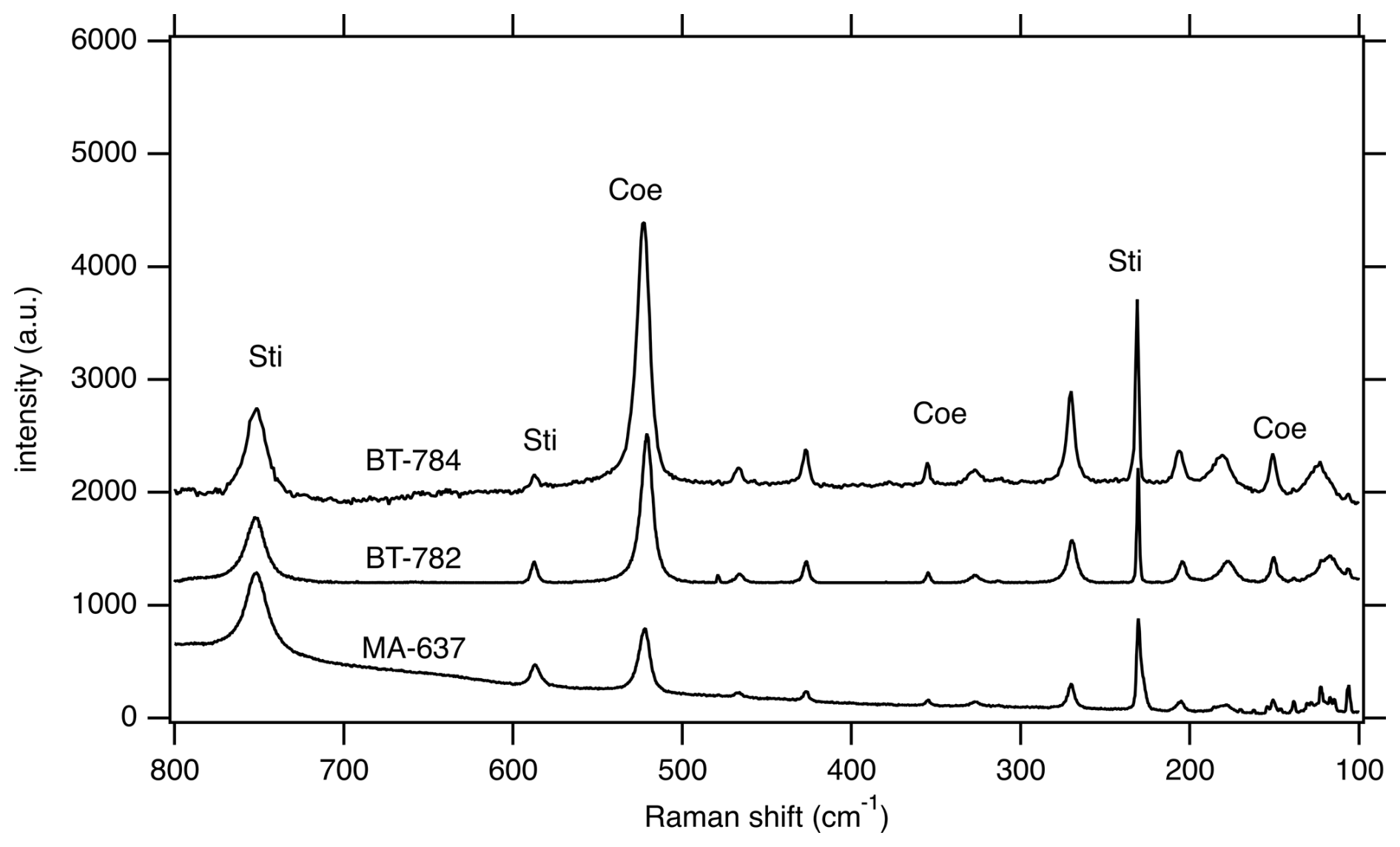
Figure 4Representative Raman spectra of samples BT-782, BT-784, and MA-637 showing mixed spectra with Sti and Coe contributions.
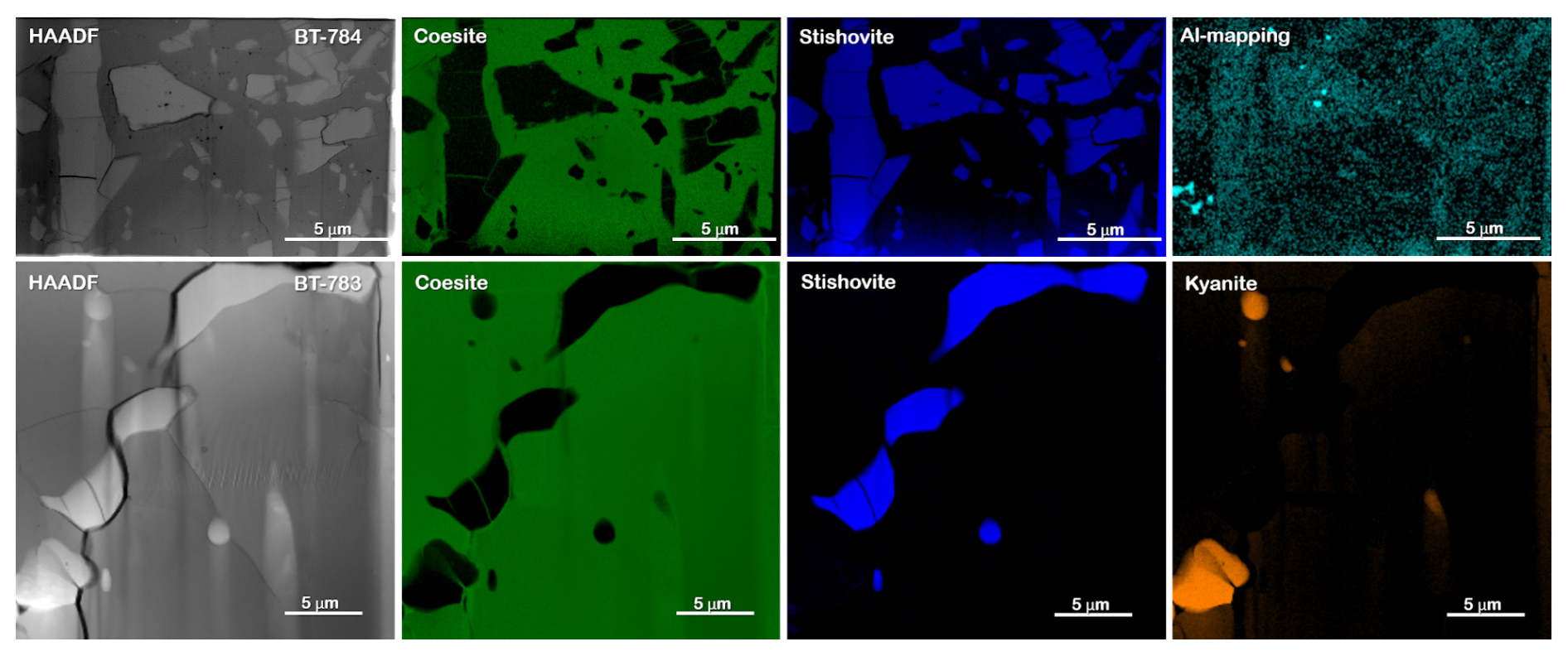
Figure 5The upper part shows the FIB foil of sample BT-784 (no. 7435) with a matrix and crystals of various sizes. Results of EELS mapping reveal that the matrix is formed by Coe (green) and the larger crystals and nanometre-sized fragments are formed by Sti (blue). Al mapping shows Al enrichment in Sti compared to Coe (greenish blue). The lower part shows details of the FIB foil of sample BT-783 (no. 7479) with worm-like nanometre-sized inclusions. Results of EELS mapping reveal that the matrix is formed by Coe, the worm-like features are formed by Sti, and the round and elongated light inclusions are formed by Ky (for details of the EELS mapping, see Fig. S4). HAADF: high-angle annular dark field.
Ex situ experiments T-016 and T-019 were carried out at 7.5 GPa and 1000 °C and 7.0 GPa and 800 °C, respectively, at relatively low pressure and thus located just below the Coe–Sti transformation line for the pure SiO2 system – in the stability field of pure Coe. In the recovered sample of T-016 mostly Coe and traces of Sti could be detected, but this was the case later only with Raman spectroscopy, while T-019 showed only Coe (Raman spectroscopy and XRPD). MA-637 with 9.6 GPa and 800 °C performed at relatively high pressure and thus located deep in the stability field of pure Sti. According to XRPD measurements the recovered sample yielded a Sti:Coe ratio of about 10:1. Figure S6a and b show a part of mount BT-637 in different magnifications. In addition to larger Sti single crystals (5–10 µm), there are regions with high porosity and an accumulation of smaller 1–2 µm sized prismatic crystals. Apart from XRPD measurements, the presence of Coe in this sample was also confirmed by Raman spectroscopy (Fig. 4), and according to electron probe mount analyses, we could perform site-specific measurements which revealed that the Coe occurrence is enriched in the porous parts. However, different to the high-temperature samples, where Sti appears darker in the BSE images than Coe, we observe in sample MA-637 no remarkable difference in the grey shade between Coe and Sti – maybe as in these samples Coe and Sti formed at relatively low temperatures, and the difference in the amount of light elements between Coe and Sti is not as large (no Z contrast) as in the high-temperature samples. The textures of low-P and low-T samples T-016 and T-019 (Fig. S6c) are quite different to that of BT-637 – they show only equally sized Coe crystals.
3.2.2 Al, Ti, and water content
The EPM Al mapping of sample BT-745 clearly visualizes that the phases involved have different Al contents with two distribution maxima. Raman spectroscopy identified only Coe in this sample, indicating that we have to deal with two different kinds of Coe crystals. EPM analyses were taken for all the experimental run products; however, due to the nanometre-sized Ky inclusions we could not fully trust these analyses. Nevertheless, they show variable Al contents, and in contrast to sample BT-745, the Al-richer phases in BT-784 and BT-783 were identified by Raman spectroscopy as Sti. The upper part of Figure S5 shows a BSE image of a part of the sample mount for BT-783 with some of the different grains, and a table included in Fig. S5 shows EMP analyses of the phases of Coe and Sti (identified by Raman spectroscopy). We selected only the most reasonable analyses (labelled 39 to 44) which nevertheless could also be falsified by Ky inclusions. The three analyses 18, 28, and 30 result in Al contents that are significantly too high due to Ky contributions. Thus, to get a better idea about the true Al contents of Coe and Sti involved, energy-dispersive spectra of both were taken from a FIB foil of sample BT-783 avoiding Ky inclusions (Fig. S5). Coe showed 0.9 wt % Al2O3, whereas Sti contained 1.3 wt % Al2O3. Thus, we conclude that all other analyses showing higher Al contents have contributions from Ky. Preferred Al incorporation into Sti compared to Coe is to be expected and also proven by the EDS Al mapping in Fig. 5. Energy-dispersive spectra of Ky were taken on FIB foils of samples BT-745, BT-783, and BT-784, and the average formula yielded Al1.98Si1.01O5 without any sign of Ti contamination.
Figure S7 shows a BSE image of the cross section of the experimental charge plus part of the wall of the Ti capsule. There is a reaction rim between the SiO2-bearing sample and the Ti. Quantification of the energy-dispersive spectra reveal about 75 wt % Ti and 25 wt % Si, corresponding to a Ti5Si3 compound. There is no evidence of Ti oxidation at these high temperatures nor Ti incorporation into the SiO2 phases. It seems that this reaction rim prevents incorporation of Ti into the SiO2 phases. EPM Ti mapping shown in Fig. S7 also shows a sharp reaction rim between the capsule and the SiO2 phases and confirms the high Ti amount in the rim and its sharpness. Numerous energy-dispersive analyses taken at either the electron probe or the TEM verify no Ti incorporation into the SiO2 phases and in kyanite.
In Fig. 6 representative FTIR spectra of Coe (T-0019) and Sti (MA-637) from experiments formed at a relatively low temperature of 800 °C are shown. They exhibit the characteristic but much broader OH bands for Coe and Sti. The quantified hydrogen incorporation is at 550 wt % ppm H2O what we expect for Sti (Fig. 6b using the calibration of Thomas et al., 2009), but it is nearly the same exceptionally high amount for Coe (Fig. 6a using the calibration of Koch-Müller et al., 2001). The broadness of the bands indicates that the hydrogen is partly disordered. Figure 6b shows the FTIR pattern for Sti (BT-783), which experienced high temperatures of 1450 °C at relatively low P before quenching – the same features (qualitatively and quantitatively) can be observed for Coe quenched from such high temperatures. Both phases do not show the characteristic IR pattern but a broad absorbance band which strongly resembles that for molecular water. This lets us suggest that the initially intrinsic OH groups are not quenchable but after (P, T) release are highly disordered in the structure and/or quenched as molecular water. Nevertheless, the broad absorbance band in the Sti spectrum of BT-783 amounts, if quantified using the calibration of Thomas et al. (2009), to about 2800 wt ppm H2O.
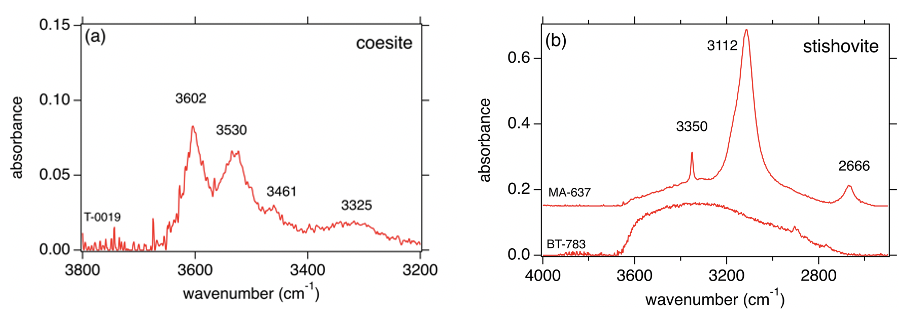
Figure 6Representative FTIR spectra of (a) Coe and (b) Sti for certain (P,T) conditions. T-0019 and MA-637 with partly ordered hydrogen in Coe and Sti, respectively, equilibrated at low temperature (800 °C); BT-783 with highly disordered hydrogen and molecular water in Sti (Fig. 6b); and Coe (same features as for Sti and therefore not shown) equilibrated at high temperature (1450 °C).
3.2.3 Powder XRD characterization of quenched solids
Results of the XRPD analyses are summarized in Table S2. In none of the analyses was kyanite detected, indicating that this phase, observed as nanometre-sized crystals by TEM, has amounts significantly less than 1 wt % in each of the samples. Due to the small amounts of materials available for XRPD and the occurrence of additional MgO reflections (contamination from material surrounding the Ti capsule), the errors in the determined cell dimensions of the SiO2 phases are rather high. Thus, the data cannot be used for further evaluation.
The Coe–Sti phase boundary is shifted in the presence of Al2O3 and H2O almost parallelly by about 1.5 GPa to lower pressures compared to results for the pure SiO2 system by Ono et al. (2017). The shift and thus the extension of the Sti stability field to lower pressure is caused by a higher Al and H incorporation into Sti compared to Coe. Higher Al content in Sti compared to Coe was proven in the recovered phases after quenching. However, it is known that the initially intrinsic hydrogen incorporation at least of stishovite is not quenchable (Spector et al., 2016; Kueter et al., 2023). In our study, in the range of the OH-stretching vibrations, both SiO2 phases formed at high temperatures (e.g. BT-783; 1450 °C) show broad absorbance bands, which strongly resemble that for molecular water and/or indicate highly disordered OH groups in their structures, while both SiO2 phases formed at low temperatures (e.g. MA-637; 800 °C) still show intrinsic OH bands. This observation is a further indication that, also in the case of Coe, the initially intrinsic OH groups are not quenchable during (P, T) release and thus the OH contents of Coe and Sti measured at room conditions underestimate those at P and T. As it is also known that the OH amount in Sti increases with T; the difference in the OH contents between Coe and Sti is even more pronounced at P and higher T. As a consequence, the Coe–Sti transition should be curved rather than be a straight line. As it is known that Sti incorporates increasing amounts of Al with increasing T (Litasov et al., 2007) and as this chemical change in Sti is presumingly more pronounced than in Coe, one can suggest that the phase boundary is curved to lower P at higher T. However, our bracketing results do not allow for a distinction, thus, even though a curved transition is thermodynamically more meaningful, for simplicity, here we present the Coe–Sti transition as a straight one.
The amount of water and Al2O3 added to SiO2 in our experiments was 10 and 1 wt %, respectively. The high water content strongly exceeds the maximum H capacity for Coe and Sti known from the literature. Thus, all experiments were performed at water-saturated conditions. However, the maximum Al capacity for Sti seems not to be reached, as some studies report higher Al contents of up to 7.6 wt % Al2O3 (Litasov et al., 2007). In our study, maximum amounts of Al concentration in Sti (coexisting with Coe) were about 1.3 wt %. This value might have been larger if a higher amount of Al had been used as the starting material for the experiments. If following the argumentation that the difference in the Al contents between Coe and Sti is responsible for the observed increase in the (P, T) field of Sti relative to Coe, then the shift in the Coe–Sti transition to lower P relative to that of the SiO2 endmembers might be even larger than the 1.5 GPa observed within this study.
In the recovered quenched samples, we observe significantly less Sti than we measured in situ by XRD before quenching. Additionally, we observed two coexisting kinds of Coe in the experiments: one relatively Al-poor Coe that, according to our interpretation, has transformed under equilibrium conditions at P–T from Sti over a certain time frame and one Al-richer Coe, sometimes pseudomorphically replacing former Sti that presumably formed during the quenching and decompression process to room conditions. This is also in line with the results of quench experiment MA-637, where we discovered in the recovered sample Coe-enriched regions with smaller 1–2 µm sized prismatic crystals (Figs. 4 and S6). Within these two types of former Sti, nanometre-sized relicts or remnants of Sti and Ky inclusions are observed by TEM (Fig. 5). Thus, during quenching, Al-rich Sti partly transforms to metastable Al-rich Coe; however, as the Al capacity to be incorporated in Coe is lower than for Sti, some Al is released and reacts with SiO2 to form nanometre-sized kyanite. At the end of each of our experiments, T is quenched to room conditions within less than a few seconds, whereas P, aimed to preserve the fragile tungsten carbide pistons intact, is released slowly within hours. The extent to which a changed quenching protocol has an influence on the partial breakdown of Sti and change of its H contents cannot be specified from the data of this study. To the best of our knowledge in none of the previous phase stability studies in the pure SiO2 system was non-quenchable stishovite reported nor discussed, not even in synthesis experiments at 9 GPa and with temperatures as low as 500 °C (Stagno et al., 2015). We attribute the non-quenchable character of the stishovite of the present study to the simultaneous incorporation of Al and H, which is supported by the results of our experiments with only Al2O3 and only H2O added to SiO2.
The observation of these changes during quenching has the following important implications. (1) Experiments on the Coe–Sti transition in a (Al, H)-bearing system would lead to erroneous results if conventional quench rather than in situ techniques at high P and T are performed. More specifically, conventional experiments would result in enlarged Coe stability and a Coe–Sti transition at much higher (P, T) conditions than determined by the in situ method. (2) As nanometre-sized kyanite in coesite are only visible by TEM, EMP analyses of Coe that have been transformed from Al-rich Sti would provide wrong Al concentrations that are too elevated.
Interestingly, our TEM observations of nanometre-sized kyanite inclusions and relicts or remnants of stishovite within coesite resemble those of Yang et al. (2007) on diamond-bearing Lubosa ophiolites (Tibet) with identical textures and phases, which were interpreted as a result of stishovite transiting back to coesite during retrograde metamorphism. According to Yang et al. (2007), “individual coesite `crystals' have external form similar to that of stishovite but are polycrystalline suggesting a pseudomorphic replacement and implying a pressure >9 GPa; some Raman spectra show small peaks consistent with remnant domains of stishovite; coesite is (in this rock) the major constituent of a kyanite-bearing silicate assemblage”. We are not aware of any further studies describing such an unusual assemblage of phases and textures in nature. The results of our study and the analogy to the study of Yang et al. (2007) might initiate further systematic investigations of similar rocks elsewhere.
Our study significantly increases the relevance of the Coe–Sti transition to be at least partly responsible for the seismic X discontinuity, because due to varying (Al, H) incorporation into the structures of the SiO2 phases, the (P, T) condition of the transition can also vary. Pugh et al. (2021) and Srinu et al. (2021) suggest that in addition to the Coe–Sti transition a second mechanism is necessary to explain the large depth variation. Pugh et al. (2021) suggest that orthoenstatite has a high-P clinoenstatite transition, while Srinu et al. (2021) are in favour of carbonate silicate melting as second mechanism. However, as shown in this study the required presence of chemical heterogeneity in explaining the depth variation of the X discontinuity could also come from variable amounts of Al and H incorporated into Sti. Compared to the pure SiO2 system the condition of our experiments results in a reduction in the transformation pressure of 1.5 GPa corresponding to a reduction at a depth of 45 km. If assuming that Sti might incorporate even larger amounts of Al at (P, T) conditions of the Coe–Sti transition than those adjusted by the starting materials in our study, the possible depth range at which the transition might occur could even be larger.
Future experiments using varying (larger) amounts of Al in the starting materials are urgently needed to verify this hypothesis. In this context, it would also be of great interest to determine if other elements (e.g. ferric iron), at which amounts, can be incorporated into Coe and Sti at the high (P, T) conditions of their transition and to which extent the transition is thereby shifted. For charge-balancing reasons, the coupled substitution Si4+ = Fe3+ + H+ is presumingly the most relevant and will have the largest effect on the shift of the Coe–Sti transition, in analogy to Al incorporation investigated in this study. To investigate reactions under the participation of water, experiments have to be performed in closed capsules. Ti capsules seem to be ideal not only because they are watertight and transparent for the synchrotron beam but also because the Ti does not react with the silicate material even at high T, due to formation of a passivating Ti5Si3 rim. Additionally, in situ methods (e.g. synchrotron XRD in multi-anvil devices or FTIR spectroscopy in diamond-anvil cells at high P and T) are absolutely required, as our and recent studies have shown that compositions or even phase ratios are changed during (P, T) quenching. Furthermore, such studies have to be undertaken with the application of techniques (e.g. TEM) in which nano-scale processes are verified, which are not seen by using conventional analytical methods (e.g. XRD or EMP). In the case of our study, without the use of TEM, the falsification of the Al contents of the SiO2 phase from nanometre-sized Ky inclusions would not have been detected.
All data derived from this research are presented in the enclosed tables, figures, and Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/ejm-36-1023-2024-supplement.
CL performed and analysed the experiments with the help of AE. MKM and BW had the initial idea for the project and prepared the paper, with the contribution of all co-authors. MKM did all the spectroscopic (FTIR and Raman) measurements. OA did the electron microprobe measurements and analyses. AS helped with the FIB preparation and collecting BSE images. SB and RF supported us during the experiments in Hamburg. VR and RW performed the TEM analyses.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This article is part of the special issue “Probing the Earth: experiments on and for our planet”. It is not associated with a conference.
The authors thank Hans-Peter Nabein and Uwe Dittmann for their technical help. Sergio Speziale, Sergey Lobanov, and Patrick O'Brien are thanked for intensive discussions. The comments of the two anonymous reviewers helped to improve the manuscript. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association (HGF), for the provision of experimental facilities. Parts of this research were carried out at beamline P61B (proposal no. I-20220780) with the support of the German Federal Ministry of Education and Research (BMBF; grant nos. 05K16WC2 and 05K13WC2). The authors thank the European Regional Development Fund and the state of Brandenburg for the Themis Z TEM (part of the Potsdam Imaging and Spectral Analysis (PISA) Facility).
The research has been supported by the Deutsche GeoForschungsZentrum. The article processing charges for this open-access publication were covered by the Helmholtz Centre Potsdam – GFZ German Research Centre for Geoscience.
The article processing charges for this open-access publication were covered by the Helmholtz Centre Potsdam – GFZ German Research Centre for Geosciences.
This paper was edited by Paola Comodi and reviewed by two anonymous referees.
Angel, R. J., Chopelas, A., and Ross, N. L.: Stability of high-density clinoenstatite at upper-mantle pressures, Nature, 358, 322–324, https://doi.org/10.1038/358322a0, 1992.
Akaogi, M., Oohata, M., Kojitani, H., and Kawaji, H.: Thermodynamic properties of stishovite by low-temperature heat capacity measurements and the coesite-stishovite transition boundary, Am. Mineral., 96, 1325–1330, https://doi.org/10.2138/am.2011.3748, 2011.
Akimoto, S.: The system MgO-FeO-SiO2 at high pressures and temperatures – phase equilibria and elastic properties, Tectonophysics, 13, 161–187, https://doi.org/10.1016/B978-0-444-41015-3.50016-1, 1972.
Bolfan-Casanova, N., Andrault, D., Amiguet, E., and Guignot, N.: Equation of state and post-stishovite transformation of Al-bearing silica up to 100 GPa and 3000 K, Phys. Earth Planet. Inter., 174, 70–77, https://doi.org/10.1016/j.pepi.2008.06.024, 2009.
Bromiley, G. D. and Pawley, A. R.: The stability of antigorite in the systems MgO-SiO2-H2O (MSH) and MgO-Al2O3-SiO2-H2O (MASH): The effects of Al3+ substitution on high-pressure stability, Am. Mineral., 88, 99–108, https://doi.org/10.2138/am-2003-0113, 2003.
Bromiley, G. D., Bromiley, F. A., and Bromiley, D. W.: On the mechanism for H and Al incorporation in stishovite, Phys. Chem. Minerals, 33, 613–621, https://doi.org/10.1007/s00269-006-0107-9, 2006.
Chen, M., Yin, C., Chen, D., Tian, L., Liu, L., and King, L.: Hydrogen solubility of stishovite provides insights into water transportation to the deep Earth, Solid Earth, 15, 215–227, https://doi.org/10.5194/se-15-215-2024, 2024.
Chen, T., Gwanmesia, G. D., Wang, X., Zou, Y., Liebermann, R. C., Michaut, X., and Li, B.: Anomalous elastic properties of coesite at high pressure and implications for the upper mantle X-discontinuity, Earth Plant. Sci. Lett., 412, 42–51, https://doi.org/10.1016/j.epsl.2014.12.025, 2015.
Chinnery, N. J., Pawley, A. R., and Clark, S. M.: In situ observation of the formation of 10Å phase from talc + H2O at mantle pressures and temperatures, Science, 286, 940–942, https://doi.org/10.1126/science.286.5441.940, 1999.
Dasgupta, R., Mallik, A., Tsuno, K., Withers, A. C., Hirth, G., and Hirschmann, M. M.: Carbon-dioxid-rich melt in the Earth's upper mantle, Nature, 493, 211–215, https://doi.org/10.1038/nature11731, 2013.
Dewaele, A., Fiquet, G., Andrault, D., and Hausermann, D.: P-V-T equation of state of periclase from synchrotron radiation measurements, J. Geophys. Res.-Solid Earth, 105, 2869–2877, https://doi.org/10.1029/1999JB900364, 2000.
Dewaele, A., Stutzmann, V., Bouchet, J., Bottin, F., Occelli, F., and Mezouar, M.: High pressure-temperature phase diagram and equation of state of titanium, Phys. Review B, 91, 134108, https://doi.org/10.1103/PhysRevB.91.134108, 2015.
Ganguly, J. and Frost, D. J.: Stability of anhydrous phase B: Experimental studies and implications for phase relations in subducting slab and the X discontinuity in the mantle, J. Geophys. Res., 111, B06203, https://doi.org/10.1029/2005JB003910, 2006.
Geisinger, K. L., Spackman, M. A., and Gibbs, G. V.: Exploring of structure, electron density distribution, and bonding in coesite with Fourier and pseudoatom refinement methods using single-crystal X-ray diffraction data, J. Phys. Chem., 91, 3237–3244, 1987.
Knapp, N., Woodland, A. B., and Klimm, K.: Experimental constraints on coesite abundances in eclogite and implications for the X seismic discontinuity, J. Geophys. Res.-Solid Earth, 120, 4910–4930, https://doi.org/10.1002/2015JB011933, 2015.
Koch-Müller, M., Fei, Y., Hauri, E., and Liu, Z.: Location and quantitative analysis of OH in coesite, Phys. Chem. Minerals, 28, 693–705, https://doi.org/10.1007/s002690100195, 2001.
Koch-Müller, M., Dera, P., Fei, Y., Reno, B., Sobolev, N., Hauri, E., and Wysoczanski, R.: OH− in synthetic and natural coesite, Am. Mineral., 88, 1436–1445, https://doi.org/10.2138/am-2003-1007, 2003.
Kroll, P., and Milko, M.: Theoretical investigation of the solid-state reaction of silicon nitride and silicon dioxid forming silicon oxynitride (Si2N2O) under pressure, Z. Anorg. Allg. Chem., 629, 1737–1750, https://doi.org/10.1002/zaac.200300122, 2003.
Kueter, N., Brugman, K., Miozzi, F., Cody, F., Yang, J., Strobel, T. A., and Walter, M. J.: Water speciation and hydrogen isotopes in hydrous stishovite: implications for the deep Earth water cycle, Contrib. Min. Petrol., 178, 48, https://doi.org/10.1007/s00410-023-02028-6, 2023.
Lakshtanov, D. L., Vanpeteghem, C. B., Jackson, J. M., Bass, J. D., Shen, G., Prakapenka, V. B., Litasov, K., and Ohtani, E.: The equation of state of Al,H-bearing SiO2 stishovite to 58 GPa, Phys. Chem. Minerals, 32, 466–470, https://doi.org/10.1007/s00269-005-0016-3, 2005.
Lakshtanov, D. L., Sinogeikin, S. V., and Bass, J. D.: High-temperature phase transitions and elasticity of silica polymorphs, Phys. Chem. Minerals, 34, 11–22, https://doi.org/10.1007/s00269-006-0113-y, 2007.
Larson, A. C. and von Dreele, R. B.: Generalized structure analysis system. Alamos National Laboratory Report LAUR 96-748, Los Alamos National Laboratory, New Mexico, 1987 pp., 2004.
Lathe, C., Koch-Müller, M., Wunder, B., Appelt, O., Bhat, S., and Farla, R.: In situ reinvestigation of reaction phase A plus high-pressure clinoenstatite to forsterite plus water in the system MgO-SiO2-H2O (MSH), Eur. J. Mineral., 34, 201–213, https://doi.org/10.5194/ejm-34-201-2022, 2022.
Li, X., Speziale, S., Glazyrin, K., Wilke, F., Liermann, H.-P., and Koch-Müller, M.: Synthesis, structure refinement and single-crystal elasticity of Al-bearing superhydrous phase B, Amer. Mineral., 107, 885–895, https://doi.org/10.2138/am-2022-7989, 2022a.
Li, X., Speziale, S., Koch-Müller, M., Husband, R. J., and Liermann, H.-P.: Phase stability of Al-bearing dense hydrous magnesium silicates at topmost lower mantle conditions: Implications for water transport in the mantle, Geophys. Res. Lett., 49, e2022GL098353, https://doi.org/10.1029/2022GL098353, 2022b.
Litasov, K. D. and Ohtani, E.: Phase relations in hydrous MORB at 18-28 GPa: implications for heterogeneity of the lower mantle, Phys. Earth Planet. Inter., 150, 239–263, https://doi.org/10.1016/j.pepi.2004.10.010, 2005.
Litasov, K. D., Kagi, H., Shatskiy, A., Ohtani, E., Lakshtanov, D. L., Bass, J. D., and Ito, E.: High hydrogen solubility in Al-rich stishovite and water transport in the lower mantle, Earth Planet. Sci. Lett., 262, 620–634, https://doi.org/10.1016/j.epsl.2007.08.015, 2007.
Liu, L.: Effects of H2O on the phase behaviour of the forsterite-enstatite system at high pressures and temperatures and implications for the Earth, Phys. Earth Planet. Int., 49, 142–167, https://doi.org/10.1016/0031-9201(87)90138-5, 1987.
Lloyd, E. C.: Editorial introduction and summary to: accurate characterization of the high-pressure environment, US Natl. Bur. Stand. Spec. Publ., 326, 1–3, 1971.
Mookherjee, M., Speziale, S., Marquardt, H., Jahn, S., Wunder, B., Koch-Müller, M., and Liermann, H.-P.: Equation of state and elasticity of the 3.65Å phase: Implications for the X-discontinuity, Am. Mineral., 100, 2199–2208, https://doi.org/10.2138/am-2015-5312, 2015.
Morishima, H., Kato, T., Urakawa, S., Utsumi, W., Suto, M., Suzuki, A., Yamazaki, D., Ohtani, E., Shimomura, O. and Kikegawa, T.: The phase boundary between α- and β-Mg2SiO4 determined by in situ X-ray observation, Science, 265, 1202–1203, 1994.
Müller, J., Koch-Müller, M., Rhede, D., Wilke, F., and Wirth, R.: Melting relations in the system CaCO3-MgCO3 at 6 GPa, Am. Mineral., 102, 2440–2449, https://doi.org/10.2138/am-2017-5831, 2017.
Nisr, C., Shim, S. H., Leinenweber, K., and Chizmeshya, A.: Raman spectroscopy of water-rich stishovite and dense high-pressure silica up to 55 GPa, Am. Mineral., 102, 2180–2189, https://doi.org/10.2138/am-2017-5944, 2017.
Ono, S., Kikegawa, T., Higo, Y., and Tange, Y.: Precise determination of the phase boundary between coesite and stishovite in SiO2, Phys. Earth Planet. Inter., 264, 1–6, https://doi.org/10.1016/j.pepi.2017.01.003, 2017.
Pamato, M. G., Myhill, R., Boffa Ballaran, T., Frost, D. J., Heidelbach, F., and Miyajima, N.: Lower-mantle reservoir implied by the extreme stability of a hydrous aluminosilicate, Nat. Geoscie., 8, 75–79, https://doi.org/10.1038/ngeo2306, 2015.
Pawley, A. R., McMillan, P. F., and Holloway, J. R.: Hydrogen in stishovite with implications of mantle water content, Science, 261, 1024–1026, https://doi.org/10.1126/science.261.5124.1024, 1993.
Piermarini, G. J. and Block, S.: Ultra high-pressure diamond-anvil cell and several semiconductor phase transition pressures in relation to fixed point pressure scale, Rev. Sci. Instrum., 46, 973–980, 1975.
Pugh, S., Jenkins, J., Boyce, A., and Cottar, S.: Global receiver function observations of the X-discontinuity reveal recycled basalt beneath hotspots, Earth Planet. Sci. Lett., 561, 116813, https://doi.org/10.1016/j.epsl.2021.116813, 2021.
Purevjav, N., Fei, H., Ishii, T., Crinit, G., Lin, Y., Mao, H.-K., and Katsura, T.: Temperature dependence of H2O solubility in Al-free stishovite, Geophys. Res. Lett., 51, e2023GL104029, https://doi.org/10.1029/2023GL104029, 2024.
Revenaugh, J. and Jordan, T. H.: Mantle layering from ScS reverberations: 3. The upper mantle, J. Geophys. Res.-Solid Earth, 96, 19781–19810, https://doi.org/10.1029/91JB01487, 1991.
Spector, K., Nylen, J., Mathew, R., Edén, M., Stoyanov, A., Leinenweber, K., and Häussermann, U.: Formation of hydrous stishovite from coesite in high-pressure hydrothermal environments, Am. Mineral., 101, 2514–2524, https://doi.org/10.2138/am-2016-5609, 2016.
Srinu, U., Kumar, P., Haldar, C., Kumar, M. R., Srinagesh, D., and Illa, B.: X-discontinuity beneath the Indian shield-Evidence for Remnant Tethyan oceanic Lithosphere in the Mantle, JGR: Solid Earth, 126, e2021JB021890, https://doi.org/10.1029/2021JB021890, 2021.
Stagno, V., Mandal, M., Landskron, K. and Fei, Y.: High pressure synthesis of mesoporous stishovite: potential applications in mineral physics, Phys. Chem. Minerals., 42, 509–515, https://doi.org/10.1007/s00269-015-0739-8, 2015.
Susaki, J., Akaogi, M., Akimoto, S., and Shimomura, O.: Garnet–perovskite transformation in CaGeO3; in-situ X-ray measurements using synchrotron radiation, Geophys. Res. Lett., 12, 729–732, 1985.
Tange, Y., Nishihara, Y., and Tsuchiya, T.: Unified analyses for P-V-T equation of state of MgO: A solution for pressure-scale problems in high P-T experiments, J. Geophys. Res., 114, B03208, https://doi.org/10.1029/2008JB005813, 2009.
Thomas, S.-M., Koch-Müller, M., Reichart, P., Rhede, D., Thomas, R., Wirth, R., and Matsyuk, S.: IR-calibration for water determination of olivine, r-GeO2, and SiO2-polymorphs, Phys. Chem. Minerals, 36, 489–509, https://doi.org/10.1007/s00269-009-0295-1, 2009.
Weidner, D. J., Bass, J. D., Ringwood, A. E., and Sinclair, W.: The single crystal moduli of stishovite, J. Geophys. Res., 87, 1334–1346, 1982.
Williams, Q. and Revenaugh, J.: Ancient subduction, mantle eclogite, and the 300 km seismic discontinuity, Geology, 33, 1–4, https://doi.org/10.1130/G20968.1, 2005.
Woodland, A. B.: The orthorhombic to high-P monoclinic transition in Mg-Fe pyroxene: Can it produce a seismic discontinuity?, Geophys. Res. Lett., 25, 1241–1244, https://doi.org/10.1029/98GL00857, 1998.
Wojdyr, M.: Fityk: a general-purpose peak fitting program, J. Appl. Crystallogr., 43, 1126–1128, https://doi.org/10.1107/S0021889810030499, 2010.
Xu, C., Inoue, T., Kakizawa, S., Noda, M., and Gao, J.: Effect of Al on the stability of dense hydrous magnesium silicate phases to the uppermost lower mantle: Implications for water transportation into the deep mantle, Phys. Chem. Minerals, 48, 31, https://doi.org/10.1007/s00269-021-01156-4, 2021.
Yagi, T. and Akimoto, S.-I.: Direct determination of coesite-stishovite transition by in-situ X-ray measurements, Tectonophysics, 35, 259–270, 1976.
Yang, J.-S., Dobrzhinetskaya, L., Bai, W.-J., Fang, Q.-S., Robinson, T. T., Zhang, J., and Green II, H. W.: Diamond- and coesite-bearing chromitites from the Luobusa ophiolite, Tibet, Geology, 35, 875–878, https://doi.org/10.1130/G23766A.1, 2007.
Zhang, J., Li, B., Utsumi, W., and Liebermann, R. C.: In situ X-ray observations of the coesite-stishovite transition: reversed phase boundary and kinetics, Phys. Chem. Minerals, 23, 1–10, https://doi.org/10.1007/BF00202987, 1996.