the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Jan 2020
| 21 Jan 2020
Adding complexity to the garnet supergroup: monteneveite, Ca3Sb5+2(Fe3+2Fe2+)O12, a new mineral from the Monteneve mine, Bolzano Province, Italy
Dan Holtstam
Luca Bindi
Paola Bonazzi
Matthias Konrad-Schmolke
Monteneveite, ideally ()O12, is a new member of the garnet supergroup (IMA 2018-060). The mineral was discovered in a small specimen belonging to the Swedish Museum of Natural History coming from the now abandoned Monteneve Pb–Zn mine in Passiria Valley, Bolzano Province, Alto Adige (South Tyrol), Italy. The specimen consists of mainly magnetite, sphalerite, tetrahedrite-(Fe) and oxycalcioroméite. Monteneveite occurs as black, subhedral crystals with adamantine lustre. They are equidimensional and up to 400 µm in size, with a subconchoidal fracture. Monteneveite is opaque, grey in reflected light, and isotropic under crossed polars. Measured reflectance values (%) at the four COM wavelengths are 12.6 (470 nm), 12.0 (546 nm), 11.6 (589 nm) and 11.4 (650 nm). The Vickers hardness (VHN100) is 1141 kg mm−2, corresponding to H=6.5–7, and the calculated density is 4.72(1) g cm−3. A mean of 10 electron microprobe analyses gave (wt %) CaO 23.67, FeO 3.75, Fe2O3 29.54, Sb2O5 39.81, SnO2 2.22, ZnO 2.29, MgO 0.15, MnO 0.03 and CoO 0.03. The crystal chemical formula calculated on the basis of a total of eight cations and 12 anions, and taking into account the available structural and spectroscopic data, is (Ca2.97Mg0.03)Σ=3.00 ()Σ=2.00()Σ=3.00O12. The most significant chemical variations encountered in the sample are related to a substitution of the type YSn4++ZFe3+→YSb5++ZFe2+. Mössbauer data obtained at RT and 77 K indicate the presence of tetrahedrally coordinated Fe2+. Raman spectroscopy demonstrates that there is no measurable hydrogarnet component in monteneveite. The six strongest Bragg peaks in the powder X-ray diffraction pattern are [d (Å), I (%), (hkl)]: 4.45, 100, (220); 3.147, 60, (400); 2.814, 40, (420); 2.571, 80, (422); 1.993, 40, (620); 1.683, 60, (642). Monteneveite is cubic, space group Ia-, with a=12.6093(2) Å, V=2004.8(1) Å3, and Z=8. The crystal structure was refined up to R1=0.0197 for 305 reflections with Fo>4σ(Fo) and 19 parameters. Monteneveite is related to the other Ca-, Sb- and Fe-bearing, nominally Si-free members of the bitikleite group, but it differs in that it is the only known garnet species with mixed trivalent and divalent cations (2:1) at the tetrahedral Z site.
Textural and mineralogical evidence suggests that monteneveite formed during peak metamorphism (at ca. 600 ∘C) during partial breakdown of tetrahedrite-(Fe) by reactions with carbonate, under relatively oxidizing conditions. The mineral is named after the type locality, the Monteneve (Schneeberg) mine.
- Article
(5062 KB) - Full-text XML
-
Supplement
(147 KB) - BibTeX
- EndNote





The garnet supergroup, with the general formula {X3} [Y2](Z3)φ12, includes all minerals isostructurally with garnet regardless of what elements occupy the four atomic sites; i.e. the supergroup includes several chemical classes (Grew et al., 2013). In particular, in recent years, several new members of the garnet supergroup that are nominally Si-free and contain elements such as Sb and Sn have been approved by the Commission on New Minerals, Nomenclature and Classification (CNMNC) of the International Mineralogical Association (IMA).
During an examination of roméite group specimens from various localities, a sample from the Monteneve (Schneeberg) mine, Passiria Valley, Bolzano Province, Alto Adige (South Tyrol), Italy (46∘53 N, 11∘ E, ∼2300 m above sea level), was found to contain a new mineral belonging to the garnet supergroup. The small sample (ca. 30 mm × 15 mm× 6 mm), labelled “Schneebergit” (iron-bearing oxycalcioroméite) (Fig. 1), had been purchased (for 15 Imperial German Goldmarks) by Hjalmar Sjögren from Dr. F. Krantz in 1897. Sjögren's collection was then donated to the Swedish Museum of Natural History in 1901.
This new member of the garnet supergroup, with the ideal formula ()O12, was named monteneveite after the type locality, and both the mineral and mineral name have been approved by the IMA CNMNC (2018–060). The name complies with the IMA nomenclature for the garnet supergroup (Grew et al., 2013), i.e. a new root name is used rather than suffixes. The holotype specimen is deposited at the Swedish Museum of Natural History, Department of Geosciences, Box 50007, SE-10405 Stockholm, Sweden, under collection number GEO-NRM catlogue no. HS3903.
The Monteneve area has been mined for its lead and zinc ores as far back as the 13th century all the way until the last mine closed in 1985, and it consists of several hundred small mines and adits (Tasser, 1994).
The ore deposits occur as bands of sulfide minerals (primarily sphalerite, pyrrhotite, galena and chalcopyrite) and are part of the Ötztal–Stubai polymetamorphic complex (ÖSC). The ores are found conformable within two distinct stratigraphic horizons of the metasedimentary Breonie metalliferous series (Frizzo et al., 1982). The Monteneve area exhibits a complex metamorphic history and some authors argue for a Cretaceous age of the metamorphism (Miller et al., 1967), but others advocate an early Alpine metamorphic age (see discussion in Sassi et al., 1985). Modern estimates of T and P on mineral assemblages in country rocks suggest peak metamorphic conditions of 600 ∘C and 8–10 kbar, respectively (Konzett et al., 2003). It is unknown from which mine level and consequently geological unit the type specimen originates; the original label simply states “Schneebergit, Schneeberg, Südtirol”. The sample could potentially be from Bockleitner Halde (“Bockleitner dump”), from where Brezina (1880) described a very similar mineral assemblage in his work on Schneebergit; however, he reported his specimen as being associated with anhydrite and gypsum, which are not found in the present material. This was later corrected by von Elterlein (1891), who pointed out that the supposed sulfates are in fact calcite. Brezina (1880) makes no mention of tetrahedrite in association with Schneebergit, even though the general description of oxycalcioroméite as 0.5 to 1 mm honey-yellow octahedra fits well with the monteneveite holotype specimen.
Monteneveite occurs mainly with magnetite, oxycalcioroméite, tetrahedrite-(Fe), sphalerite and chalcopyrite (Figs. 2 and 3). Sphalerite frequently contains minute inclusions of chalcopyrite. The average composition of tetrahedrite-(Fe) (by means of five energy-dispersive X-ray spectroscopy, EDS, point analyses) is Cu9.5Ag0.1(Fe1.4Zn0.6)Sb4.1S12.9. Oxycalcioroméite occurs as 0.1–1 mm honey-yellow to orange-brown euhedral crystals, and the composition is Ca1.4Fe0.4Sb1.8Sn0.2O7 (mean of eight EDS point analyses). Magnetite occurs both as euhedral crystals and as anhedral grains enveloped by monteneveite.
In addition, the sample contains minor amounts of anthophyllite, gudmundite and genplesite; the latter represents the second recorded occurrence after that from the Talnakh Cu–Ni deposit, Norilsk, Russian Federation (Pekov et al., 2018). The sample is quite vuggy, but we suspect that it has been treated with dilute acid at some point in order to dissolve the carbonates (mainly calcite) to reveal the oxycalcioroméite crystals.
Monteneveite forms subhedral crystals, with a maximum size of up to about 0.4 mm and essentially equidimensional in shape. The grains are commonly fractured, with radial patterns. The macroscopic colour is black (opaque), with brownish black streak and adamantine lustre. The crystals are non-fluorescent and non-magnetic. Microintendation hardness was measured with a Shimadzu M-type hardness tester, yielding VHN100=1072–1246, with an average of 1141 kg mm−2 (11.2 GPa) from four indentations, corresponding to a Mohs hardness of 6.5–7. There is no observed cleavage or parting; the fracture is subconchoidal. The density could not be empirically determined due to paucity of material. Using the empirical formula, the calculated density is 4.72(1) g cm−3 for single-crystal X-ray diffraction data.

Figure 1Macroscopic photograph of monteneveite (mnv) in the holotype specimen, GEO-NRM catalogue no. HS3903, here seen together with almost euhedral, honey-yellow crystals of oxycalcioroméite and associated minerals. Field of view is 2.5 mm. © Lorin. All rights reserved.
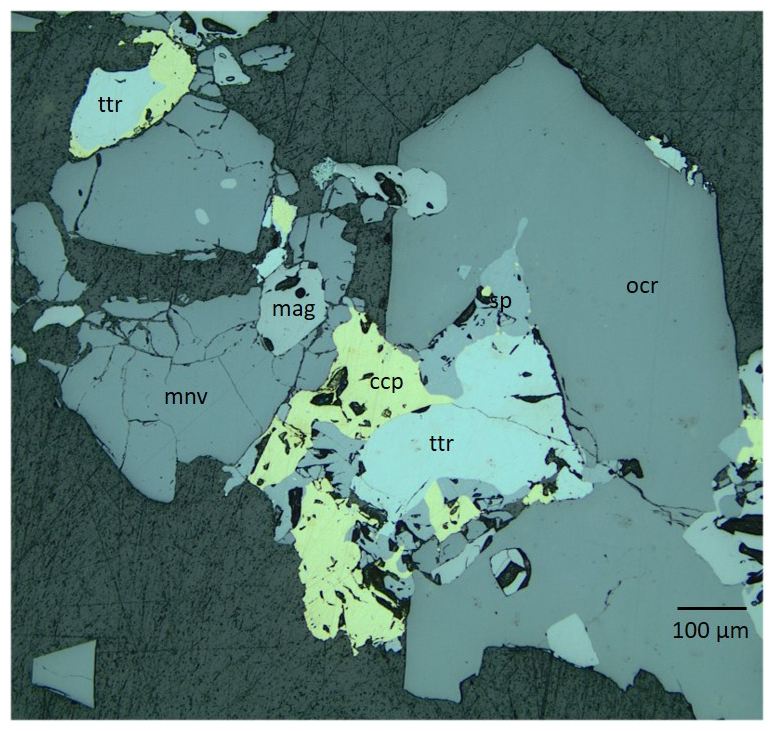
Figure 2Reflected-light microscope picture illustrating the textural relations between the sulfides and oxides. Blue-filtered incandescent light. Abbreviations: mnv – monteneveite; mag – magnetite; ocr – oxycalcioroméite; ttr – tetrahedrite-(Fe); sp – sphalerite; ccp – chalcopyrite. Sample GEO-NRM catalogue no. HS3903.

Figure 3Field-emission scanning electron microscope image (BSE) of monteneveite and associated phases. The image shows the same section as in Fig. 2 (with some rotation). Abbreviations: mnv – monteneveite; mag – magnetite; ocr – oxycalcioroméite; ttr – tetrahedrite-(Fe); sp – sphalerite; ccp – chalcopyrite. Sample GEO-NRM catalogue no. HS3903.
Table 1Reflectance data for monteneveite. Interpolated values for COM (Commission of Ore Minerals) wavelengths are given in bold.
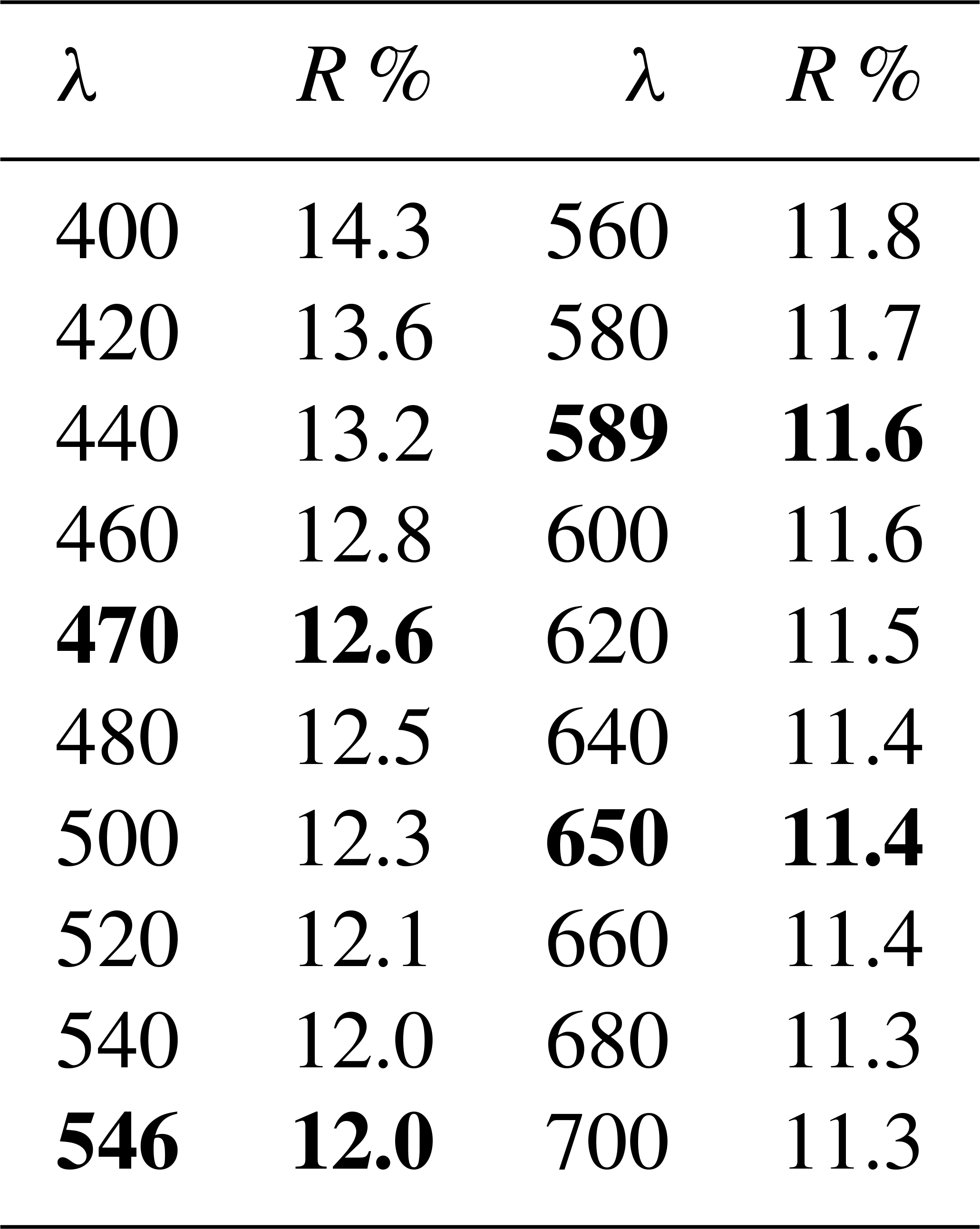
Table 2Chemical composition (wt %) of monteneveite from EPMA analyses.

* is calculated to obtain overall electroneutrality in the formula.
In reflected-light microscopy, the colour is grey (Fig. 2). The overall reflectivity is significantly lower than that of associated magnetite, but similar to that of oxycalcioroméite. The latter mineral, in contrast to monteneveite, shows many internal reflections. Monteneveite is isotropic under crossed polars. Reflectance data were measured in air with an AVASPEC-ULS2048 × 16 spectrometer attached to a Zeiss Axiotron UV microscope (10 Ultrafluar objective), using a halogen lamp (100 W) and a SiC (Zeiss no. 846) standard, with a measured field of 150 µm in diameter. The results (average of 300 scans, integration time 30 ms) are given in Table 1.
The chemical composition of monteneveite was determined using a JEOL JXA-8200 electron probe microanalyser (EPMA), fitted with five wavelength dispersive spectrometers, at the Institute of Earth and Environmental Sciences of Potsdam University. The instrument was operated at a working distance of 10 mm with an accelerating voltage of 15 kV and a beam current of 15 nA. The beam size was 2 µm. Counting times were 20 s for major element peaks and 10 s left and right of the peak in order to determine background counts during measurement. The elements were calibrated on the following reference materials: Fe (almandine), Zn (willemite), Sn (cassiterite), Sb (stibnite), Co (pentlandite), Mn (bustamite), Mg (almandine) and Ca (andradite). All standards were from Astimex except for andradite (Smithsonian). Na, Si, Al, Ti, Zr and Cu were sought but not detected.
The analytical results obtained on the same crystal used for single-crystal X-ray diffraction (see below) are given in Table 2 (average of 10 point analyses). The empirical formula was calculated on the basis of eight cations, and the partition was calculated to obtain overall electroneutrality. The chemical data reveal that in monteneveite the Sb5+ (and Fe2+) contents exhibit negative covariance with respect to Sn4+ (and Fe3+), thus confirming that a dzhuluite-type substitution occurs in the sample.
Taking the results from structural and spectroscopic investigation into account, the crystal chemical formula is the following: (Ca2.97Mg0.03)Σ=3.00()Σ=2.00 ()Σ=3.00O12. The analytical totals, which are consistently higher than 100 %, are in accord with the absence of H2O (see results from Raman spectroscopy) and other volatiles. The ideal formula is {Ca3}[]()O12, which requires CaO 23.26, Sb2O544.73, Fe2O322.08, FeO 9.93 (total 100.00 wt %).
The chemical composition of some minerals associated with monteneveite was determined using an FEI Quanta 650 field-emission scanning electron microscope, fitted with an 80 mm2 X-MaxN Oxford Instruments EDS detector hosted at the Department of Geosciences, Swedish Museum of Natural History, Stockholm, Sweden (20 kV, beam size 1 µm, working distance 10 mm).
Laser-induced Raman measurements were carried out at room temperature using a Horiba (Jobin Yvon) LabRAM HR Evolution hosted at the Department of Earth Sciences, University of Gothenburg, Gothenburg, Sweden. The samples were excited with an air-cooled frequency-doubled 532 nm Nd:YAG laser utilizing an Olympus 100× objective (numerical aperture = 0.9). The lateral resolution of the unpolarized confocal laser beam was of the order of 1 µm. Spectra were generated in the range of 100 to 4000 cm−1 utilizing a 600 grooves cm−1 grating and a thermoelectric cooled electron multiplier charge-coupled device (CCD) including a front illuminated 1600×200 pixel chip. The spectral resolution on unoriented polished crystals of monteneveite embedded in epoxy resin was of the order of 1 cm−1. The wavenumber calibration was performed using the 520.7 cm−1 Raman band on a polished silicon wafer with a wavenumber accuracy usually better than 0.5 cm−1. Raman spectra of the sample were collected through five acquisition cycles with single counting times of 45 s in a near-back-scattered geometry.
The Raman spectrum of monteneveite (Fig. 4) is similar to that of the minerals in the bitikleite group (in particular dzhuluite; Galuskina et al., 2013a). There is a strong band at 729 cm−1 related to symmetric stretching vibrations of the tetrahedral Fe3+–O bond; the broadening of this band could be attributed to the co-presence of Fe2+ at the Z site as also inferred from Mössbauer spectroscopy (see below). The medium-intensity band at 586 cm−1 could be caused by asymmetric stretching vibrations in the (Fe3+O4)5− tetrahedra but is shifted to a lower wavenumber than for the related minerals bitikleite and usturite (Galuskina et al., 2010b). Similarly, the strong band at 485 cm−1 related to bending vibrations of tetrahedral Fe3+–O is also shifted to lower wavenumbers. Bands at lower wavenumbers (at 269 cm−1 and the nearby right shoulder) may be attributed to translational motion of the tetrahedral Z position and translational motions of Ca–O in the X position. There are no obvious bands that belong to symmetric or asymmetric stretching vibrations of Sb5+–O units; according to Guillén-Bonilla et al. (2014), who investigated synthetic byströmite-type CoSb2O6, there should be manifestation of stretching and coupling at ∼518 cm−1, asymmetric stretching at ∼617 cm−1 and symmetric stretching at ∼691 cm−1. None of these features could be identified with the exception of the last one, which may correspond to the shoulder of the 729 cm−1 band in monteneveite. Additionally, Bahfenne and Frost (2010) reported roméite-group minerals to have a strong band from 505 to 518 cm−1 related to the symmetric stretching mode of octahedrally coordinated Sb, but this feature is absent in the Raman spectra of monteneveite, or possibly quite weak and therefore obscured in the shoulder by the 485 cm−1 band. There are no bands visible in the OH-stretching vibration region (3000–3800 cm−1). Therefore, we have concluded that there is no significant hydrogarnet component (O4H4) that could contribute to charge balance.
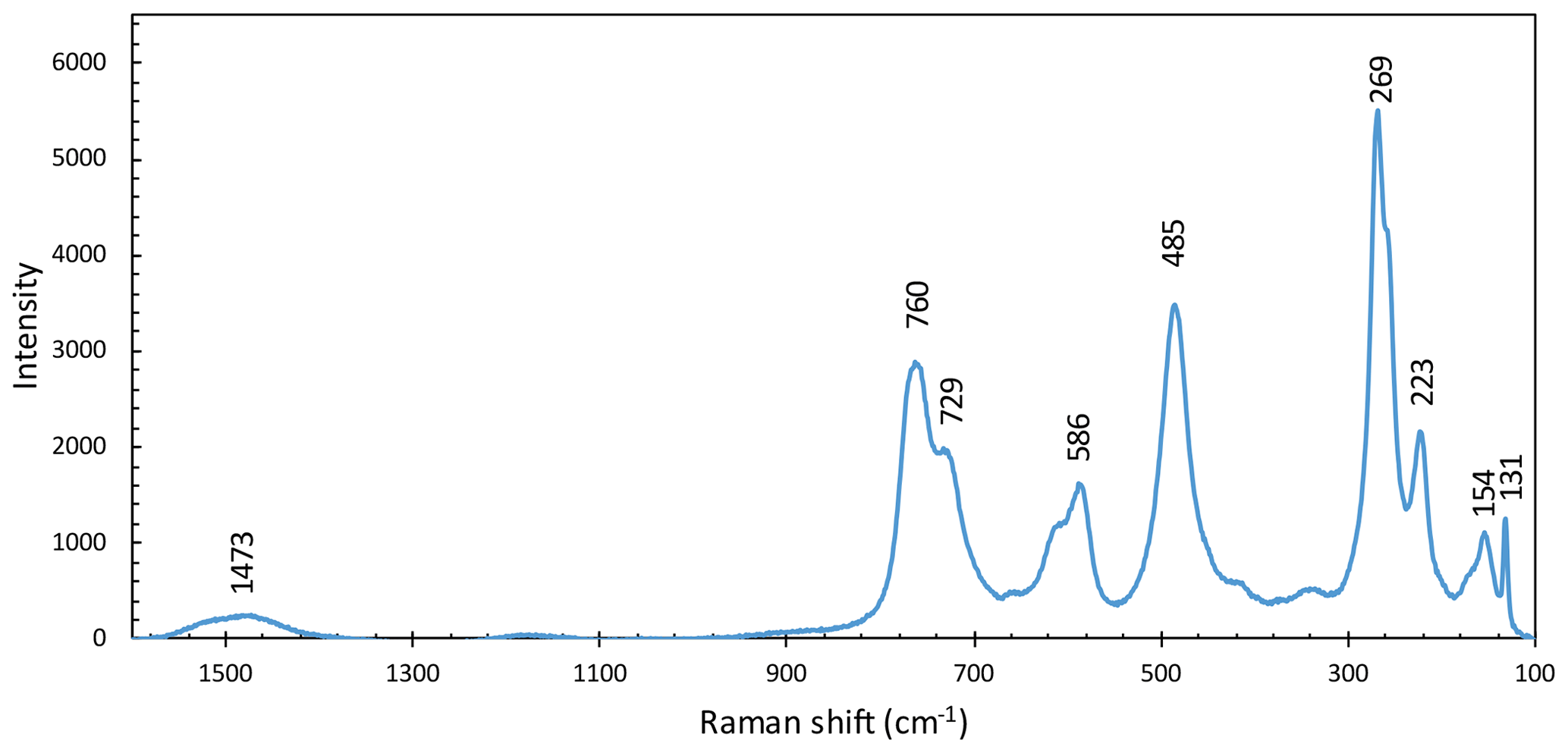
Figure 4Raman spectrum of monteneveite from 1600 to 100 cm−1 with the most prominent peaks indicated.
About 40 small (diameter ranging from 50 to 300 µm) crystals of monteneveite were handpicked from a crushed rock chip and mounted on carbon tape in order to validate their identity with SEM-EDS analysis; they were then turned over and their chemistry checked again to make sure the Mössbauer spectra would be essentially free from other phases. The crystals were ground in an agate mortar and mixed with thermoplastic resin and pressed into a 13 mm tablet. The experiments were carried out employing a conventional spectrometer system (hosted at the Department of Geosciences, Swedish Museum of Natural History, Stockholm, Sweden) operated in constant-acceleration mode with a 57Co γ-radiation source (nominally 1.8 GBq). The spectra were collected in 54.7∘ geometry at 298 and 77 K (with a liquid-N2 cryostat), during 2 weeks each over 1024 channels covering the velocity range from −4.2 to +4.2 mm s−1. Spectrum calibration was performed against Fe foil, and analysis was carried out assuming Lorentzian line shapes with the MossA program (Prescher et al., 2012). Relative absorption areas and hyperfine parameters obtained for monteneveite in the final fitting procedure are given in Table 3.
Table 3Mössbauer hyperfine parameters for monteneveite, at 298 and 77 K.
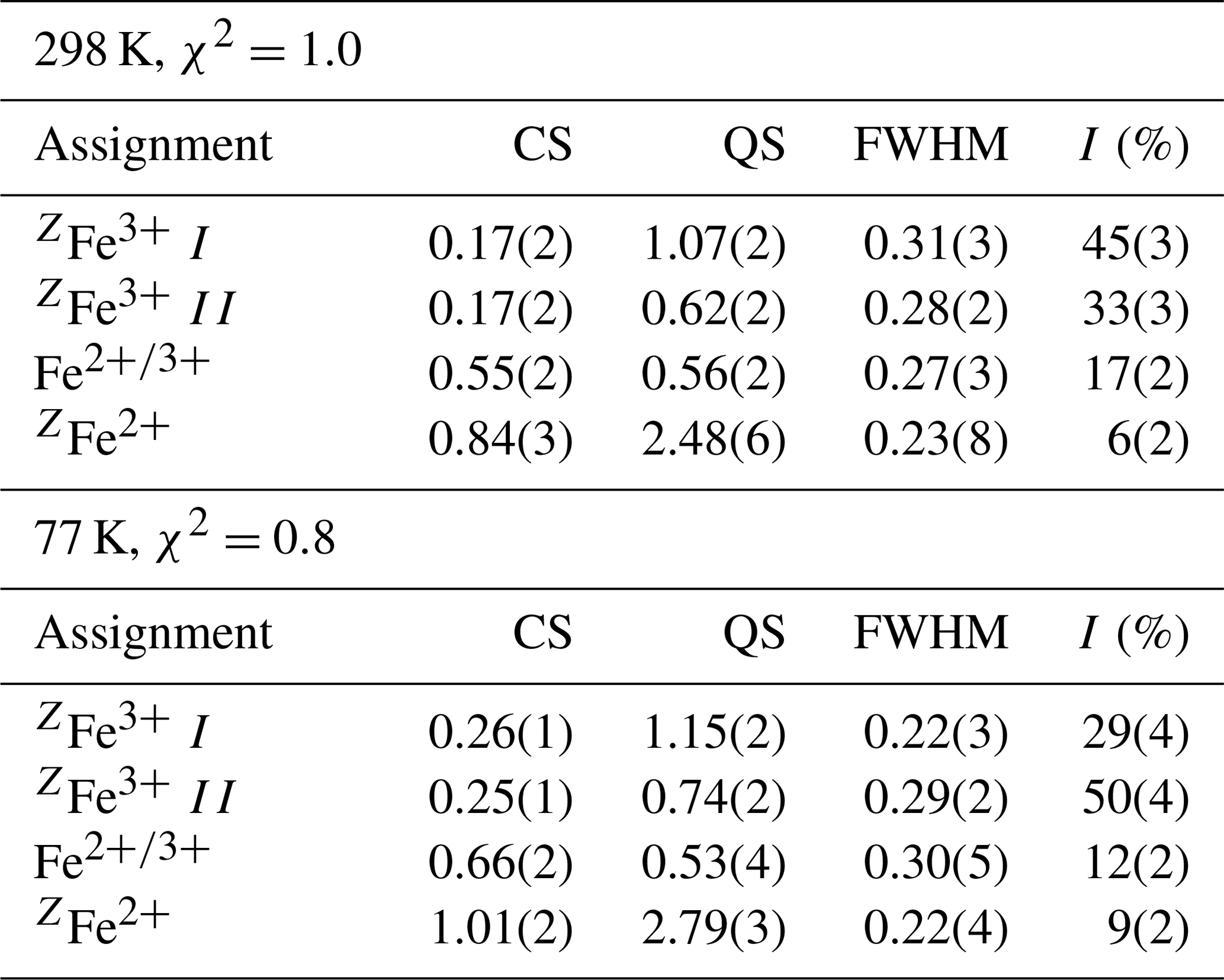
Notes: CS (centre shift), QS (quadrupole splitting), and FWHM (full width at half maximum) given in millimetres per second, and I is area as a percentage of the total Fe at that site and valency.
Both spectra (Fig. 5a, b) are dominated by a doublet with a broad absorption centred around 0.17 (298 K) and 0.25 (77 K) mm s−1. This feature is assigned to the dominant Fe3+ at the tetrahedrally coordinated Z site, based on the hyperfine parameters, in full agreement with numerous Mössbauer studies of natural (mostly schorlomite; Amthauer et al., 1977; Schwartz et al., 1980; Wu and Mu, 1986; Locock et al., 1995) and Fe3+- and Sb5+-bearing synthetic (Berry et al., 1996) garnet samples. To deal with the line broadness (ca. 0.40 mm s−1 FWHM), a distribution of the quadrupole splitting is assumed, in practice treated by introducing a pair of doublets in the fitting. This approach was also used by Berry et al. (1996) for the subspectrum in synthetic {YCa2}[](Fe3+)3O12.
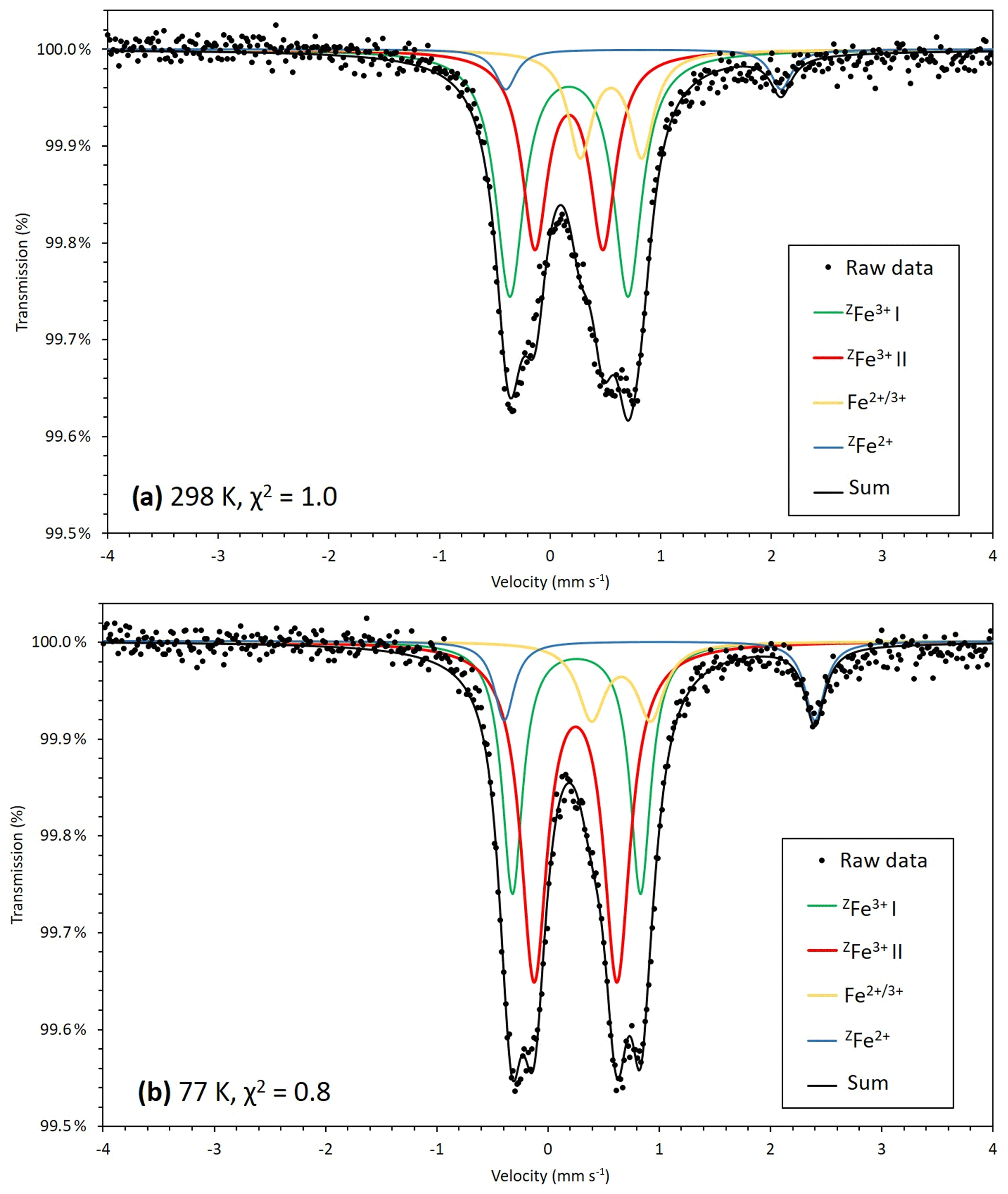
Figure 5Transmission 57Fe Mössbauer spectrum of monteneveite measured at 298 K, χ2=1.0 (a) and 77 K, χ2=0.8 (b). Both green and red lines correspond to [4]Fe3+ while [4]Fe2+ is represented by the blue line. The yellow line cannot directly be attributed to either octahedrally coordinated Fe3+ or Fe2+ and may represent a delocalized electronic state, which could explain why the 77 K spectrum is better resolved.
Of the two spectra of monteneveite collected, the one at 77 K (Fig. 5b) is the most well resolved. Specifically, a relatively weak doublet is evident by its absorption on the high-velocity side, and it is fitted to be centred at ∼1.0 mm s−1 and with a large quadrupole splitting (∼2.8 mm s−1). We interpret this as originating from the tetrahedrally coordinated Fe2+ (blue line in Fig. 5b). The parameters are in good agreement with those found for some natural garnet samples at cryogenic temperatures (Locock et al., 1995; Chakhmouradian and McCammon, 2005) and generally for Fe2+ occupying regular tetrahedra in oxides (e.g. Larsson et al., 1994; Holtstam, 1996). In the present spectra, the appearance of this peak is clearly temperature-dependant, as it is less protruding in the 298 K spectrum (Fig. 5a) and therefore more difficult to constrain. In addition to these two absorption features, a relatively weak doublet with small quadrupole splitting needs to be introduced in the fitting. It has an intermediate centroid shift value (0.66 mm s−1 at 77 K) that cannot be attributed with confidence to either octahedrally coordinated Fe3+ or Fe2+. With respect to garnet structures, data from the literature are not homogenous, with centroid shifts (at low temperatures) usually higher than 1.2 mm s−1 and lower than 0.50 for six-coordinated Fe2+ and Fe3+ (Amthauer et al., 1977; Wu and Mu, 1986; Locock et al., 1995; Chakmouradian and McCammon, 2005). In fact, the value here is more characteristic for a delocalized electronic state, corresponding to a charge intermediate between the formal 2+ and 3+. A charge-transfer process involving [4]Fe2+ and [6]Fe3+ could explain this feature and agrees well with the observed temperature behaviour; thermally induced electron hopping decreases, and hence we see the [4]Fe2+ doublet develop towards lower temperatures at the expense of the absorption related to the delocalized state of Fe valence electrons (Table 3).
There is no general consensus that intervalence charge transfer occurs in garnet structures for Fe2+ and Fe3+ hosted in corner-sharing tetrahedra and octahedra, although it is has been inferred from Mössbauer data in some studies (Huggins et al., 1977; Wu and Mu, 1986; Chakmouradian and McCammon, 2005). It should be noted that the amount of tetrahedrally coordinated Fe3+, calculated to 82 % of total Fe from the EPMA-based formula, shows a good agreement with band area ratios from the Mössbauer data, 79(4) and 78(3) % at 77 and 298 K, respectively. Considering the charge-transfer phenomena, it would not be possible to exactly calculate the amount of Fe2+, but the fraction obtained at 77 K (9 %) is not too far from the expected value (12 %). This approach also assumes similar recoil-free fractions for Fe2+ and Fe3+ at the different sites, which may be only approximately true.
7.1 Powder X-ray diffraction data
X-ray powder diffraction data (Table 4) were obtained on the fragment used for the single-crystal study (see below) with an Oxford Diffraction Xcalibur PX Ultra diffractometer fitted with a 165 mm diagonal Onyx CCD detector and using copper radiation (CuKα, λ=1.54138 Å). The working conditions were 40 kV and 40 nA with 1 h of exposure; the detector-to-sample distance was 7 cm. The program CrysAlis RED was used to convert the observed diffraction rings to a conventional powder diffraction pattern. Least-squares refinement gave the following unit-cell values: a=12.6032(2) Å and V=2001.9(1) Å3.
Table 4Measured and calculated X-ray powder diffraction data (d in Å) for monteneveite. The strongest diffraction lines are given in bold.
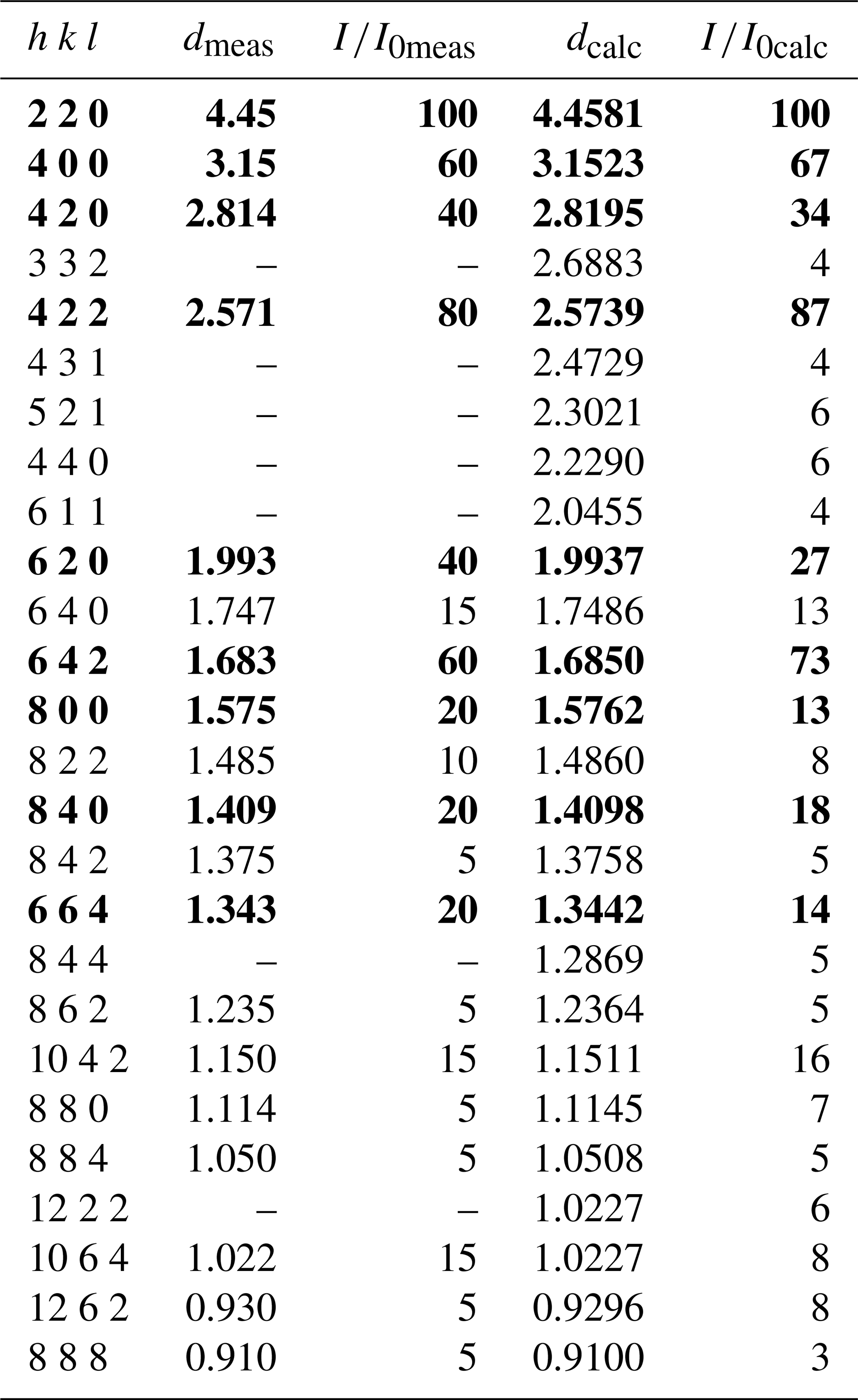
Note: calculated diffraction pattern obtained with the atomic parameters reported in Table 5 (only reflections with are listed).
7.2 Single crystal X-ray diffraction data
A single fragment of monteneveite (0.065 mm × 0.110 mm× 0.180 mm) was examined with an Oxford Diffraction Xcalibur single-crystal diffractometer equipped with a CCD detector, with graphite-monochromatized MoKα radiation (λ=0.71073 Å). The collected data were integrated and corrected for standard Lorentz and polarization factors with the CrysAlis RED package (Oxford Diffraction, 2006). The software ABSPACK in CrysAlis RED (Oxford Diffraction, 2006) was used for the absorption correction. Table 5 reports details of the selected crystal, intensity data collection and refinement.
Table 5Data and experimental details for the selected monteneveite crystal.
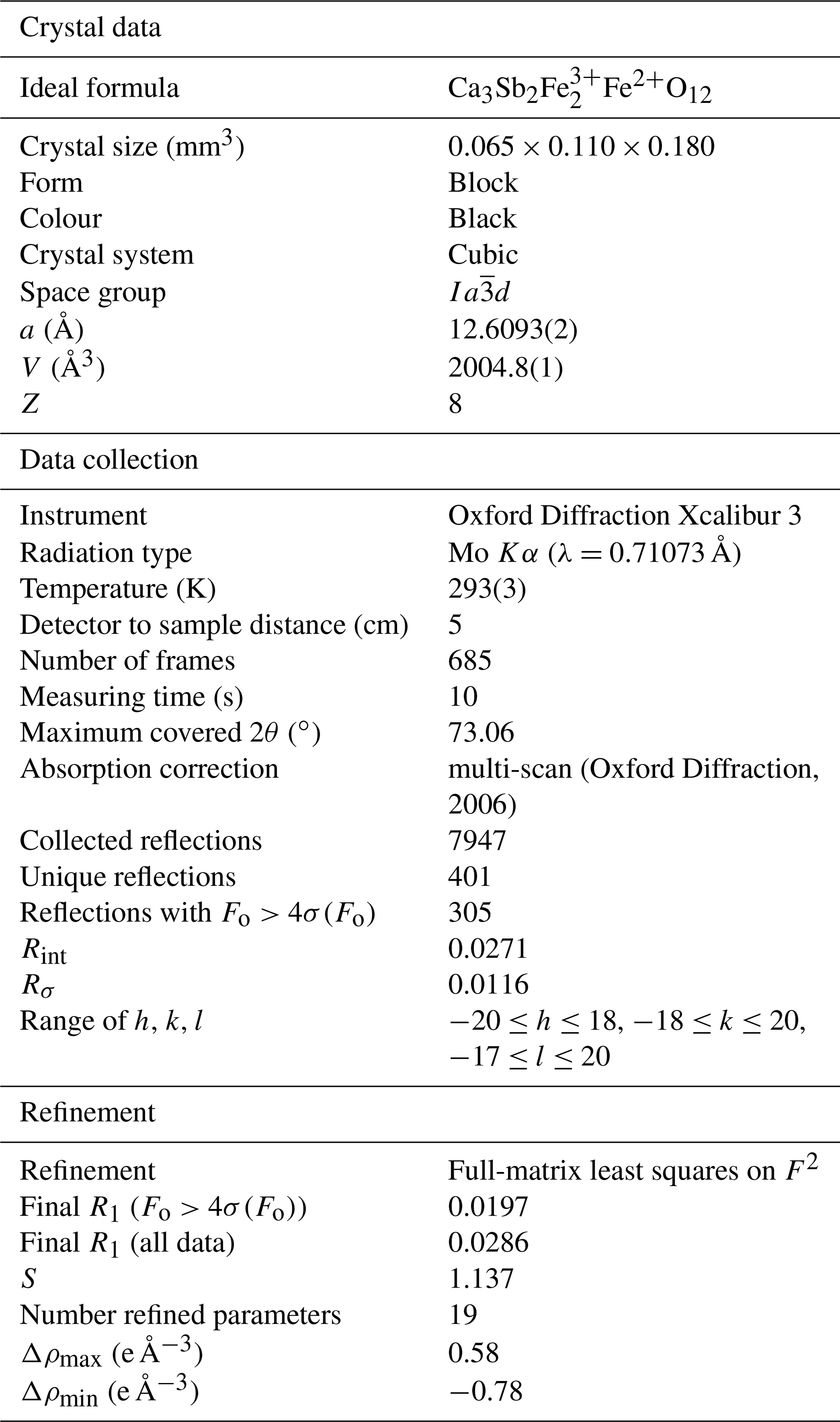
Deviations from the cubic symmetry of the unit-cell values were well below their standard deviation. A cubic unit cell [a=12.6093(2) Å] was assumed and the intensity data were merged according to the Laue group (Rint=2.71 %). The crystal structure was refined in the starting from the atomic coordinates of andradite (Hazen and Finger, 1989). Scattering curves for neutral atoms were taken from the International Tables for Crystallography (Wilson, 1992). During the first refinement cycles, the occupancy at the X and Z sites was allowed to vary (Ca vs. □ and Fe vs. □). The refined mean electron numbers were 20.0(2) and 26.0(2) e− for the X and Z sites, respectively, and therefore their site occupancy factors (sof's) were fixed in the subsequent stages of the refinement. However, the sof at the Y site was allowed to vary (Sb vs. Fe). After several cycles of anisotropic refinement, a final R1=0.0197 for 305 reflections with Fo>4σ(Fo) with 19 refined parameters was achieved (0.0286 for all 401 reflections). Atomic coordinates, site occupancies and equivalent isotropic displacement parameters are given in Table 6. Selected bond distances and bond-valence sums (calculated according to the parameters of Brese and O'Keeffe, 1991) are given in Table 7.
Table 6Atoms, site occupancy factors (sof), fractional atomic coordinates and equivalent isotropic displacement parameters (Å2) for the selected monteneveite crystal.

Table 7Bond distances (Å) and bond valence sums (BVS in v.u.) in the crystal structure of monteneveite.

8.1 Crystal chemistry
The crystal structure of monteneveite (Fig. 6) is identical to that of the cubic members of the garnet supergroup (Grew et al., 2013). The overall chemical formula, as obtained through the single-crystal X-ray diffraction study is Ca3[Sb0.93Fe0.07]2Fe3O12 (Z=8), in excellent agreement with that obtained from EPMA data (overall mean electron number from X-ray data = 236.5; overall mean electron number from chemical data = 236.4). Such a cation distribution is in agreement with the observed bond distances. The minor amounts of Sn4+ at the octahedrally coordinated site (Y) and Zn at the tetrahedrally coordinated site (Z) were assigned on the basis of the preference of these cations shown in toturite or irinarassite (Galuskina et al., 2010a, 2013b) and yafsoanite (Mills et al., 2010), respectively. The tetrahedron shows a mean Z–O distance of 1.888(1) Å, slightly longer than that expected for pure Fe3+–O, which, on the basis of EXAFS measurements, was estimated to be close to 1.85 Å (Giuli et al., 2012); on the other hand, < Z-O> in monteneveite is shorter than the corresponding value (1.905 Å) observed in the synthetic garnet (Bhim et al., 2017), in keeping with a content of tetrahedral divalent cations less than 1.00 atoms pfu. The presence of measurable quantities of divalent Fe at the Z site in a garnet structure is not too common, but it has been demonstrated with spectroscopic methods, both for synthetic material (e.g. Antonini et al., 1981) as well as in natural samples (Amthauer et al., 1977; Locock et al., 1995).
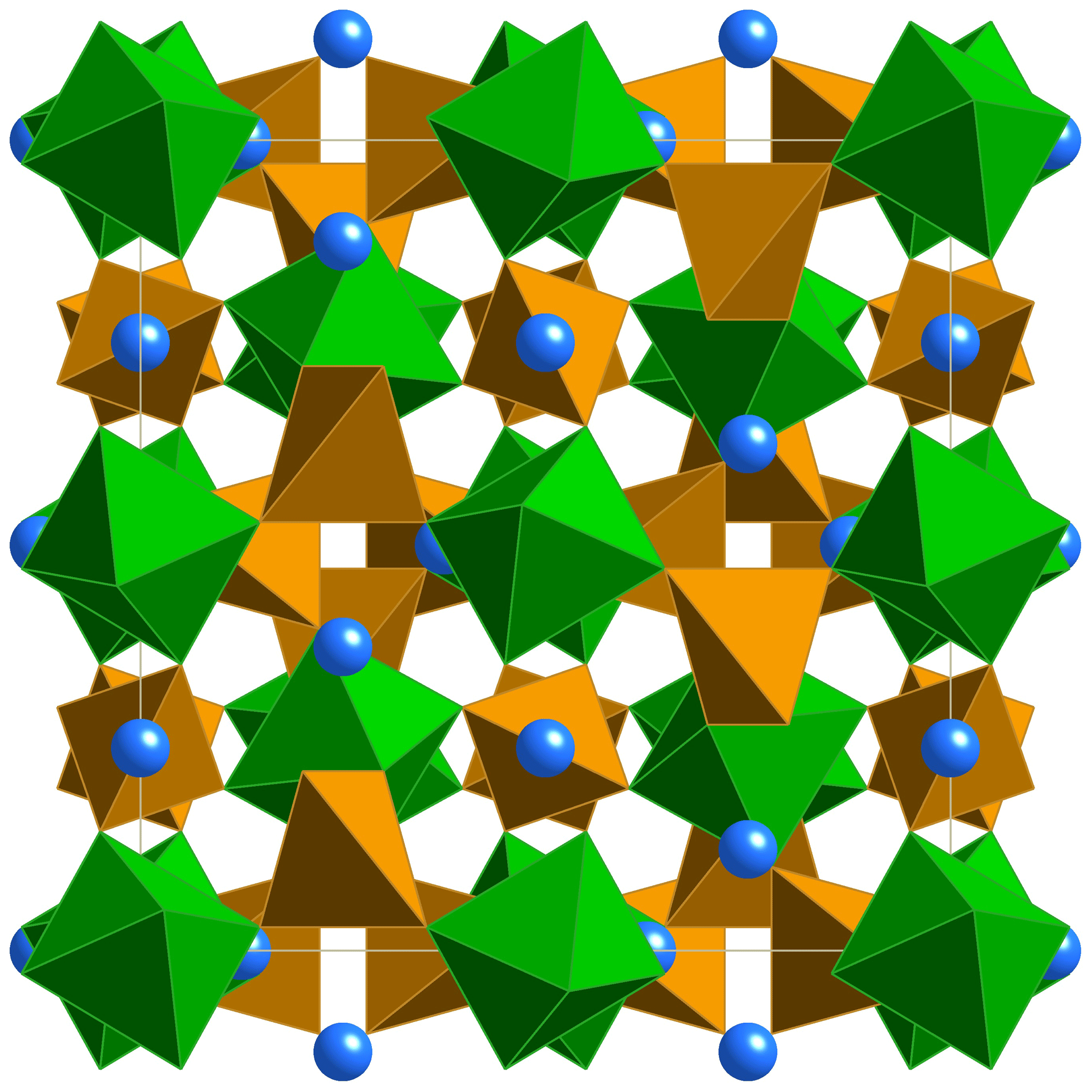
Figure 6The monteneveite crystal structure seen along [100]. Blue spheres– X site (Ca); green octahedra– Y site (Sb); brown tetrahedra– Z site (Fe).
The dominance of Sb5+ at the six-coordinated Y site is in good agreement with the mean bond distance [1.990(1) Å], slightly larger than the pure octahedral <Sb5+-O> distances from literature: 1.9647 Å in roméite (Matsubara et al., 1996), 1.960–1.983 Å in ingersonite (Bonazzi and Bindi, 2007), 1.958 Å in filipstadite (Bonazzi et al., 2013) and 1.971–2.022 Å in rinmanite (Holtstam et al., 2001). Due to the very large volume of the tetrahedron, the X dodecahedron (which shares two edges with it) shows a mean distance (2.490 Å) much larger than that observed, for example, in andradite (2.425 Å, X= Ca, Z= Si3; Hazen and Finger, 1989), or even in schorlomite (2.440 Å, X= Ca, Z= ; Peterson et al., 1995). For the same reason, its degree of distortion is very low [Δ(X–O) = 0.144 Å].
It can be noted that the microhardness of monteneveite is only slightly lower than that of andradite (11.2 vs. 11.5 GPa), which is in agreement with the general picture that the larger the volume of the XO8 polyhedron, the higher the compressibility of the garnet structure (Milman et al., 2001). As an example, grossular, which shows an <X–O> of 2.403 Å (Hazen and Finger, 1978), exhibits a higher value of microhardness (13.2 GPa).
Monteneveite is chemically related to dzhuluite, {Ca3} [Sb5+Sn] (Galuskina et al., 2013a), according to the substitution vector YSb5+ + ZFe2+ → YSn4+ + ZFe3+ and to usturite, {Ca3}[Sb5+Zr]()O12 (Galuskina et al., 2010b), via YSb5+ + ZFe2+ → YZr +Z Fe3+. The present sample has a conspicuous component (33 %) of an unaccredited garnet species of end-member composition {Ca3} []()O12, achieved through the substitution 0.5 YSb5+ + ZFe2+ → 0.5 YFe3+ + ZFe3+, and a significant fraction (20 %) of the hypothetical Zn analogue of monteneveite {Ca3}[](Zn)O12 via the substitution vector ZFe2+ → ZZn2+.
Table 8Comparison of monteneveite to other related species in the garnet supergroup.
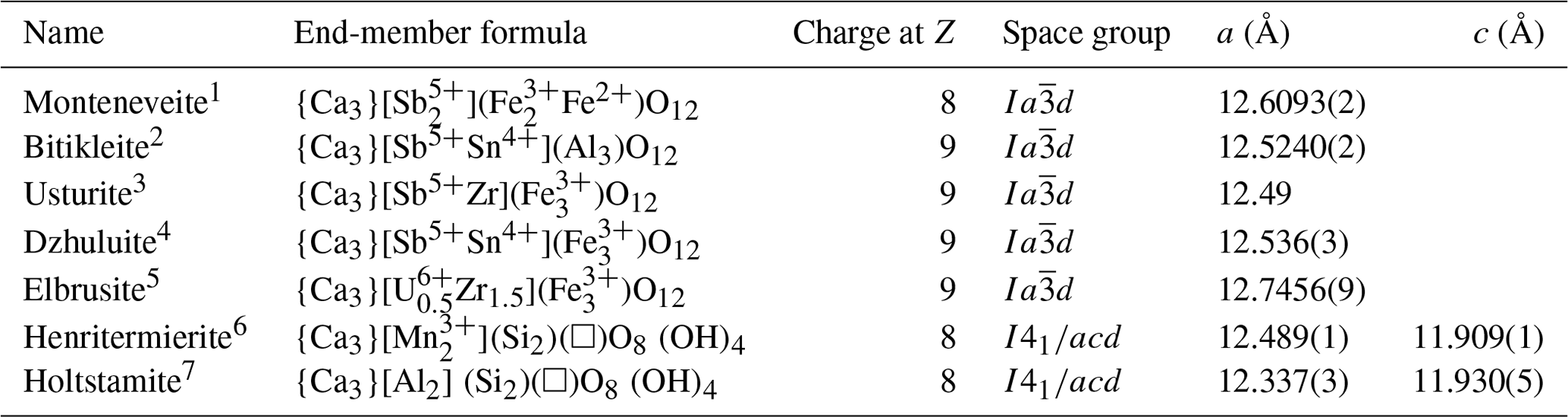
1 This work. 2 Galuskina et al. (2010b). 3 Galuskina et al. (2010b). 4 Galuskina et al. (2013a). 5 Galuskina et al. (2010c). 6 Armbruster et al. (2001). 7 Hålenius et al. (2005).
8.2 Remarks on nomenclature
Monteneveite, ideally {Ca3}[]()O12, belongs to the garnet supergroup (Grew et al., 2013) and is so far the only known member of the garnet supergroup with nominal mixed ( population at the tetrahedral Z site. The other garnet minerals having a total charge at Z site = 8 are henritermierite (Armbruster et al. 2001) and holtstamite (Hålenius et al., 2005), which, however, achieve the total charge by a mixed () population. On the mere basis of total charge at the Z site, monteneveite should be classified into the henritermierite group. Nonetheless, the crystallochemical properties strongly differ from those of the henritermierite group minerals on some critical points:
-
monteneveite is cubic, whereas henritermierite and holtstamite crystallize in the tetragonal system, space group I41/acd;
-
the general formula is CaO12 instead of Ca)O12;
-
monteneveite is not a silicate.
Furthermore, in spite of the chemical similarities (see Table 8) and identical symmetry (space group ), monteneveite does not fit with the bitikleite group (charge at Z site = 9). For these reasons and because the subdivision of a group into subgroups is strongly discouraged (Grew et al., 2013), it seems appropriate to introduce a new group – provided that other distinct members of this type are discovered (e.g. {Ca3}[](Zn)O12). In a general mineral classification (Nickel–Strunz), monteneveite belongs to the oxide class, subdivision 4.CC.
8.3 Origin of monteneveite
Monteneveite is most likely a product of skarn formation during regional metamorphism. The partial breakdown of tetrahedrite-(Fe) and recrystallization of calcite would generate the chemical constituents needed to form both monteneveite and oxycalcioroméite. Large parts of the sulfide assemblages were probably formed during early diagenetic and low-metamorphic conditions. At peak metamorphism these were resorbed and partially recrystallized. The source of Sn is unknown, but the concentrations observed in monteneveite and oxycalcioroméite might originally have been mobilized from the partial dissolution of stannite, not observed in the present sample but reported from the deposit by Mair et al. (2007). Mineralogical evidence suggests that fO2 was within the magnetite stability field since magnetite is present both as idiomorphic crystals contiguous to monteneveite and as inclusions. It is probable that upper-amphibolite-facies metamorphic conditions (∼600 ∘C and 8–10 kbar) would be required in order to form monteneveite, which is further favoured by a Si- and Al-poor local environment. To the best of our knowledge, monteneveite is the first completely Si-free natural garnet species (not considering the fluoride cryolithionite) at the detection level of EPMA used in our study (∼0.035 wt %) compared to what is reported in the literature; the amount is even less than the 0.07±0.04 wt % SiO2 found for a specimen of manganberzeliite (Nagashima and Armbruster, 2012).
All data needed to draw the conclusions in the present study are shown in this paper. The CIF is available in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/ejm-32-77-2020-supplement.
AK found the mineral and performed the initial chemical analyses. AK and MKS collected micro-Raman data, and AK and DH interpreted the spectrum. LB and PB did the single-crystal X-ray diffraction experiment and refined the crystal structure. AK and DH performed Mössbauer spectroscopy and interpreted the data. MKS collected electron-microprobe data and AK interpreted the results. AK and DH wrote the paper with contributions from all coauthors.
The authors declare that they have no conflict of interest.
Special thanks to Christina Günter, University of Potsdam, for her help with the EPMA work and to Torbjörn Lorin for the colour photograph of the specimen. Additional thanks go to Henrik Skogby for help with preparing the sample for Mössbauer spectroscopy. Luca Bindi and Paola Bonazzi thank CRIST, Laboratorio di Cristallografia Strutturale, University of Florence, Italy. This paper benefited from reviews by E. Galuskin and the anonymous journal reviewer.
This paper was edited by Sergey Krivovichev.
Amthauer, G., Annersten, H., and Hafner, S. S.: The Mössbauer spectrum of 57Fe in titanium-bearing andradites, Phys. Chem. Miner., 1, 399–413, 1977.
Antonini, B., Geller, S., Paoletti, A., Paroli, P., and Tucciarone, A.: Site occupancy of ferrous ions in iron garnets, J. Magn. Magn. Mater., 22, 203–206, 1981.
Armbruster, T., Kohler, T., Libowitzky, E., Friedrich, A., Miletich, R., Kunz, M., Medenbach, O., and Gutzmer, J.: Structure, compressibility, hydrogen bonding, and dehydration of the tetragonal Mn3+ hydrogarnet, henritermierite, Am. Mineral., 86, 147–158, 2001.
Bahfenne, S. and Frost, R. L.: Raman spectroscopic study of the antimonate mineral roméite, Spectrochim. Acta A, 75, 637–639, 2010.
Berry, F. J., Dávalos, J. Z., Gancedo, J. R., Greaves, C., Marco, J. F., Slater, P., and Vithal, M.: Cation distribution and magnetic interactions in substituted iron-containing garnets: characterization by iron-57 Mössbauer spectroscopy, J. Solid State Chem., 122, 118–129, 1996.
Bhim, A., Gopalakrishnan, J., Laha, S., and Natarajan, S.: Color tuning in garnet oxides: The role of tetrahedral coordination geometry for 3 d metal ions and ligand–metal charge transfer (Band-gap manipulation), Chem.-Asian J., 12, 2734–2743, 2017.
Bonazzi, P. and Bindi, L.: The crystal structure of ingersonite, , and its relationships with pyrochlore, Am. Mineral., 92, 947–953, 2007.
Bonazzi, P., Chelazzi, L., and Bindi, L.: Superstructure, crystal chemistry, and cation distribution in filipstadite, a Sb5+-bearing, spinel-related mineral, Am. Mineral., 98, 361–366, 2013.
Brese, N. E. and O'Keeffe, M.: Bond-valence parameters for solids, Acta Crystallogr. B, 47, 192–197, 1991.
Brezina, A.: Über ein neues Mineral, den Schneebergit. Verhandlungen der kaiserlich-königlichen Reichsanstalt Wien, 17, 313–314, 1880.
Chakhmouradian, A. R. and McCammon, C. A.: Schorlomite: a discussion of the crystal chemistry, formula, and inter-species boundaries, Phys. Chem. Miner., 32, 277–289, 2005.
Frizzo, P., Mills, J., and Visona, D.: Ore petrology and metamorphic history of Zn-Pb ores, Monteneve, Tyrol, N. Italy, Miner. Dep., 17, 333–347, 1982.
Galuskina, I. O., Galuskin, E. V., Dzierzanowski, P., Gazeev, V. M., Prusik, K., Pertsev, N. N., Winiarski, A., Zadov, A. E., and Wrzalik, R.: Toturite Ca3Sn2Fe2SiO12–A new mineral species of the garnet group, Am. Mineral., 95, 1305–1311, 2010a.
Galuskina, I. O., Galuskin, E. V., Armbruster, T., Lazic, B., Dzierzanowski, P., Gazeev, V. M., Prusik, K., Pertsev, N. N., Winiarski, A., Zadov, A. E., Wrzalik, R., and Gurbanov, A. G.: Bitikleite-(SnAl) and bitikleite-(ZrFe): new garnets from xenoliths of the Upper Chegem volcanic structure, Kabardino-Balkaria, Northern Caucasus, Russia, Am. Mineral., 95, 959–967, 2010b.
Galuskina, I. O., Galuskin, E. V., Armbruster, T., Lazic, B., Kusz, J., Dzierzanowski, P., Gazeev, V. M., Pertsev, N. N., Prusik, K., Zadov, A. E., Winiarski, A., Wrzalik, R., and Gurbanov, A. G.: Elbrusite-(Zr) –a new uranian garnet from the Upper Chegem caldera, Kabardino-Balkaria, Northern Caucasus, Russia, Am. Mineral., 95, 1172–1181, 2010c.
Galuskina, I. O., Galuskin, E. V., Kusz, J., Dzierżanowski, P., Prusik, K., Gazeev, V. M., Pertsev, N. N., and Dubrovinsky, L.: Dzhuluite, , a new bitikleite-group garnet from the Upper Chegem Caldera, Northern Caucasus, Kabardino-Balkaria, Russia, Eur. J. Mineral., 25, 231–239, 2013a.
Galuskina, I. O., Galuskin, E. V., Prusik, K., Gazeev, V. M., Pertsev, N. N., and Dzierżanowski, P.: Irinarassite Ca3Sn2SiAl2O12–new garnet from the Upper Chegem Caldera, Northern Caucasus, Kabardino-Balkaria, Russia, Mineral. Mag., 77, 2857–2866, 2013b.
Giuli, G., Cicconi, M. R., and Paris, E.: The [4]Fe3+-O distance in synthetic kimzeyite garnet, Ca3Zr2[Fe2SiO12], Eur. J. Mineral., 24, 783–790, 2012.
Grew, E. S., Locock, A. J., Mills, S. J., Galuskina, I. O., Galuskin, E. V., and Hålenius, U.: Nomenclature of the garnet supergroup, Am. Mineral., 98, 785–811, 2013.
Guillén-Bonilla, A., Rodríguez-Betancourtt, V. M., Flores-Martínez, M., Blanco-Alonso, O., Reyes-Gómez, J., Gildo-Ortiz, L., and Guillén-Bonilla, H.: Dynamic response of CoSb2O6 trirutile-type oxides in a CO2 atmosphere at low-temperatures, Sensors, 14, 15802–15814, 2014.
Hålenius, U., Häussermann, U. and Harryson, H.: Holtstamite, Ca3(Al, Mn3+)2 (SiO4)3−x (H4O4)x, a new tetragonal hydrogarnet from Wessels Mine, South Africa, Eur. J. Mineral., 17, 375–382, 2005.
Hazen R. M. and Finger L. W.: Crystal structures and compressibilities of pyrope and grossular to 60 kbar, Am. Mineral., 63, 297–303, 1978.
Hazen R. M. and Finger L. W.: High-pressure crystal chemistry of andradite and pyrope: Revised procedures for high-pressure diffraction experiments, Am. Mineral., 74, 352–359, 1989.
Holtstam, D.: Iron in hibonite: a spectroscopic study. Phys. Chem. Miner., 23, 452–460, 1996.
Holtstam, D., Gatedal, K., Söderberg, K., and Norrestam, R.: Rinmanite, Zn2Sb2Mg2Fe4O14(OH)2, a new mineral species with a nolanite-type structure from the Garpenberg Norra mine, Dalarna, Sweden, Can. Mineral., 39, 1675–1683, 2001.
Huggins, F. E., Virgo, D., and Huckenholz, H. G.: Titanium-containing silicate garnets. II. The crystal chemistry of melanites and schorlomites, Am. Mineral., 62, 646–665, 1977.
Konzett, J., Hoinkes, G., and Tropper, P.: Alpine metamorphism in the Schneeberg Complex and neighbouring units (immediate vicinity of Obergurgl), In 5th Workshop of Alpine Geological Studies, Field Trip Guide E, Geol. Paläont. Mitt. Innsbruck, 26, 21–45, 2003.
Larsson, L., O'Neill, H. S. C., and Annersten, H.: Crystal chemistry of synthetic hercynite (FeAl2O4) from XRD structural refinements and Mössbauer spectroscopy, Eur. J. Mineral., 6, 39–52, 1994.
Locock, A., Luth, R. W., Cavel, R. G., Smith, D. G. W., and Duke, M. J. M.: Spectroscopy of the cation distribution in the schorlomite species of garnet, Am. Mineral., 80, 27–38, 1995.
Mair, V., Vavtar, F., Schölzhorn, H., and Schölzhorn, D.: Der Blei-Zink-Erzbergbau am Schneeberg, Südtirol, Mitt. Österr. Miner. Ges., 153, 145–180, 2007.
Matsubara, S., Kato, A., Shimizu, M., Sekiuchi, K., and Suzuki, Y.: Romeite from the Gozaisho mine, Iwaki, Japan, Mineral. J., 18, 155–160, 1996.
Miller, D. S., Jäger, E., and Schmidt, K.: Rb-Sr-Altersbestimmungen an Biotiten der Raibler-Schichten des Brenner Mesozoikums und am Muskovitgranitgneis von Vent (Ötztaler Alpen), Eclogae Geol. Helv., 60, 537–541, 1967.
Mills, S. J., Kampf, A. R., Kolitsch, U., Housley, R. M., and Raudsepp, M.: The crystal chemistry and crystal structure of kuksite, Pb3Zn3Te6+P2O14, and a note on the crystal structure of yafsoanite, (Ca,Pb)3Zn(TeO6)2, Am. Mineral., 95, 933–938, 2010.
Milman, V., Akhmatskaya, E.V., Nobes, R. H., Winkler, B., Pickard, C. J., and White, J. A.: Systematic ab initio study of the compressibility of silicate garnets, Acta Crystallogr., B57, 163–177, 2001.
Nagashima, M. and Armbruster, T.: Palenzonaite, berzeliite, and manganberzeliite: (As5+, V5+, Si4+) O4 tetrahedra in garnet structures, Mineral. Mag., 76, 1081–1097, 2012.
Oxford Diffraction: CrysAlis RED (Version 1.171.31.2) and ABSPACK in CrysAlis RED. Oxford Diffraction Ltd, Abingdon, Oxfordshire, England, 2006.
Pekov, I. V., Sereda, E. V., Zubkova, N. V., Yapaskurt, V. O., Chukanov, N. V., Britvin, S. N., Lykova, I. S., and Pushcharovsky, D. Y.: Genplesite, Ca3Sn(SO4)2(OH)6 ⋅ 3H2O, a new mineral of the fleischerite group: first occurrence of a tin sulfate in nature, Eur. J. Mineral., 30, 375–382, 2018.
Peterson, R. C., Locock, A. J., and Luth, R. W.: Positional disorder of oxygen in garnet: the crystal-structure refinement of schorlomite, Can. Mineral., 33, 627–631, 1995.
Prescher, C., McCammon, C., and Dubrovinsky, L.: MossA: a program for analyzing energy-domain Mössbauer spectra from conventional and synchrotron sources, J. Appl. Crystallogr., 45, 329–331, 2012.
Sassi, F. P., Cavazzini, G., Visona, D., and Del Moro, A.: Radiometric geochronology in the Eastern Alps: results and problems, Rend. Soc. It. Mineral. Petrol., 40, 187–224, 1985.
Schwartz, K. B., Nolet, D. A., and Burns, R. G.: Möessbauer spectroscopy and crystal chemistry of natural Fe–Ti garnets, Am. Mineral., 65, 142–153, 1980.
Tasser, R.: Das Bergwerk am Südtiroler Schneeberg: Landesbergbaumuseum, Verlag-Anst, Athesia, 1994.
von Elterlein, A.: Beiträge zur Kenntniss der Erzlagerstätte des Schneebergs bei Mayrn in Südtirol, Jahrbuch der Geologischen Bundesanstalt, 41, 289–348, 1891.
Wilson, A. J. C. (Ed.): International Tables for Crystallography: Mathematical, Physical, and Chemical Tables (Vol. 3), International Union of Crystallography, 1992.
Wu, G. and Mu, B.: The crystal chemistry and Mössbauer study of schorlomite, Phys. Chem. Miner., 13, 198–205, 1986.
- Abstract
- Introduction
- Occurrence and paragenesis
- Appearance and physical properties
- Chemical composition
- Raman spectroscopy
- Mössbauer spectroscopy
- X-ray crystallography
- Results and discussion
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Occurrence and paragenesis
- Appearance and physical properties
- Chemical composition
- Raman spectroscopy
- Mössbauer spectroscopy
- X-ray crystallography
- Results and discussion
- Data availability
- Author contributions
- Competing interests
- Acknowledgements
- Review statement
- References
- Supplement