the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Sep 2020
| 11 Sep 2020
Theoretical infrared spectra of OH defects in corundum (α-Al2O3)
Etienne Balan
The atomic-scale structure, relative stability and infrared spectroscopic properties of OH defects in corundum (α-Al2O3) are theoretically investigated at the density functional theory level. Comparison with experimental data makes it possible to assign most of the narrow bands observed between 3150 and 3400 cm−1 in natural and Ti- or V-doped synthetic corundum to specific defects. These defects correspond to the association of one OH group with an Al vacancy and M4+ for Al3+ substitutions in neighboring sites. The OH group is located in the large oxygen triangle forming the base of the vacant Al site. Models of interstitial proton associated with a nearby Mg2+ for Al3+ substitution are consistent with the broad band observed at 3010 cm−1 in Mg-doped corundum. Its is also suggested that two weaker OH-stretching bands observed in nominally pure synthetic corundum at 3163 and 3209 cm−1 could be associated with intrinsic defects combining an Al and an O vacancy. These results highlight the importance of defect clustering in the high-temperature incorporation of hydrogen in nominally anhydrous minerals.
- Article
(5052 KB) - Full-text XML
-
Supplement
(398 KB) - BibTeX
- EndNote





Corundum (α-Al2O3) is an important refractory mineral which forms in a large variety of natural environments ranging from the primitive solar system to the Earth lithosphere (e.g., Bowles et al., 2011). Natural gem-quality ruby and sapphire, whose color is related to Cr or Fe and Ti impurities, are emblematic corundum varieties of cultural and trading importance (e.g., Muhlmeister et al., 1998; Smith, 1995; Rossman, 2009). As a nominally anhydrous mineral, corundum is known to incorporate some amount of hydrogen under the form of structural OH groups, which are readily observed using infrared spectroscopy (e.g., Eigenmann and Günthard, 1971; Volynets et al., 1972; Beran, 1991; Smith, 1995; Wöhlecke and Kovács, 2001; Libowitzky and Beran, 2006). The hydrogen concentration is however very low, typically below 0.5 wt. ppm of H2O in natural corundum (Beran and Rossman, 2006). Corundum is also an oxide ceramic with important industrial uses related to its mechanic, dielectric and optic properties. To this respect, occurrence of trace quantities of hydrogen in alumina deserves particular attention because it can affect the transport properties, mechanical strength and response to irradiation of the material (e.g., Engstrom et al., 1980; Ramírez et al., 1997a, b, 2004; Kronenberg et al., 2000).
The infrared spectra of synthetic or natural corundum often display a combination of well-resolved OH-stretching bands between 3150 and 3400 cm−1 (Eigenmann and Günthard, 1971; Volynets et al., 1972; Beran, 1991; Moon and Phillips, 1991, 1994; Kronenberg et al., 2000; Ramírez et al., 1997a, b, 2004; Libowitzky and Beran, 2006). All these bands are pleochroic with a dominant polarization parallel to the (001) plane (Engstrom et al., 1980; Moon and Phillips, 1994; Ramírez et al., 2004). Experimental studies of natural and synthetic corundum have suggested that the prominent bands at 3310, 3230 and 3185 cm−1 are associated with titanium ions occurring in the corundum structure as a tetravalent chemical impurity (Moon and Phillips, 1991, 1994). Protons bound to oxygen atoms and structural tetravalent cations substituted in nearby Al sites are expected to locally compensate for the electrostatic charge deficit due to an aluminum vacancy. Importantly, the clustering scheme of tetravalent impurities around the Al vacancy is expected to modify the OH-stretching frequency. Following Moon and Phillips (1991, 1994), the 3310 cm−1 band would be related to OH defects associated with two Ti4+ ions while the 3230 and 3185 cm−1 bands would correspond to association with a single Ti4+ ion. Relative OH band intensities have thus been used to infer Ti diffusion properties and clustering equilibria as a function of temperature in α-Al2O3. Weaker bands are also observed at 3209, 3296 and 3366 cm−1 (Kronenberg et al., 2000; Ramírez et al., 2004). In a V-doped synthetic sample, prominent bands are observed at 3183.9, 3229.4, 3278.3 and 3291.5 cm−1, and a weaker band is observed at 3382 cm−1 (Ramírez et al., 2004). The relative intensity of the OH-stretching bands in Ti- and V-doped samples experimentally treated at high temperature (1400 K) depends on their cooling rate (Ramírez et al., 2004), which is consistent with the record of a temperature-dependent distribution of OH defects displaying different clustering schemes. In addition, a broader band extending from 2900 to 3100 cm−1 with a dominant polarization in the direction perpendicular to the (001) plane is observed in Mg-doped samples (Volynets et al., 1972; Ramírez et al., 1997b). Broad bands with similar properties have been reported at 3025 and 2972 cm−1 in Co- and Ni-doped samples, respectively (Eigenmann and Günthard, 1971).
Although these experimental studies brought strong constraints for the interpretation of OH-stretching infrared spectra of corundum, a full picture in terms of local geometry of the defects has not been obtained. In addition, some of the above-listed bands are observed in nominally “pure” synthetic samples (e.g., Turner and Crawford, 1975; Engstrom, 1980; Ramírez et al., 2004), and their relation to specific impurities or defects occurring at very low concentration levels may prove to be uncertain. To this end, a theoretical approach linking the microscopic structure of the defects to their spectroscopic properties could be useful, as attested by theoretical investigations of OH groups in nominally anhydrous silicates (e.g., Wright, 2006; Balan et al., 2017, 2020; Blanchard et al., 2017; Jollands et al., 2020). Previous theoretical studies have focused on the association of hydrogen with intrinsic defects in corundum (Zhang et al., 2014; T-Thienprasert et al., 2017). Based on theoretical vibrational frequencies, T-Thienprasert et al. (2017) concluded that the OH-stretching bands observed at ∼3200–3300 cm−1 in corundum were consistent with the association of H and Al vacancies.
In the present study, the microscopic structure of a series of defect models associating H atoms with clustered Al vacancies and Ti4+ or V4+ cations is theoretically determined, and their spectroscopic properties are compared to available experimental data. The results confirm the dominance of this type of defects in the infrared spectra of natural and synthetic corundum and shed light on the nature of some debated bands.
The properties of OH defects in corundum were theoretically investigated using the same approach and numerical parameters as in Jollands et al. (2020) and Balan et al. (2020). The defect modeling was performed within the density functional theory (DFT) framework using the generalized gradient approximation (GGA) to the exchange-correlation functional as proposed by Perdew, Burke and Ernzerhof (PBE functional; Perdew et al., 1996). The modeling scheme used periodic boundary conditions and a plane-wave basis set as implemented in the PWscf code of the Quantum Espresso package (Giannozzi et al., 2009; http://www.quantum-espresso.org, last access: 8 September 2020). Ionic cores were described using optimized norm-conserving Vanderbilt (ONCV) pseudopotentials (Hamann, 2013; Schlipf and Gygi, 2015), and a plane-wave energy cutoff of 80 Ry was used, ensuring a convergence of the total energy better than 1 mRy per atom.
Structural properties of OH-bearing defects were determined using corundum supercells (120 atoms) containing up to three hydrogen atoms. Brillouin zone sampling for the electronic integration was restricted to the Γ point. Unit-cell parameters of pure corundum (S.G. were optimized at zero pressure ( Å, c=13.08 Å) and were used without further relaxation to produce the OH-bearing supercells. As usually observed in DFT modeling performed at the GGA level, the theoretical cell lengths are overestimated with respect to their experimental counterparts ( Å, c=12.991 Å; Newnham and de Haan, 1962). For all systems, the relaxation of atomic internal coordinates was performed until the residual forces were Ry per atomic unit. For the models of electrostatically charged defects, a compensating homogeneous electrostatic background was spread over the supercell to ensure the macroscopic neutrality of the periodic system.
The vibrational modes at the Brillouin zone center (Γ point), the Born effective charge tensors and the electronic dielectric tensor were calculated using the linear response theory (Baroni et al., 2001) as implemented in the PHonon code (Giannozzi et al., 2009; http://www.quantum-espresso.org, last access: 8 September 2020). The high-frequency OH stretching modes that are decoupled from the other vibrational modes occurring at significantly lower frequency can be accurately calculated by only considering the displacement of the oxygen and hydrogen atoms involved in OH groups (Balan et al., 2008). The complex low-frequency dielectric permittivity tensor has been calculated for each defect by adding only the ionic contributions related to the OH-stretching modes to the electronic permittivity tensor and using an arbitrary damping parameter of 4 cm−1, accounting for the width of absorption bands (Balan et al., 2008). For isolated OH groups, the theoretical angle between the [001] direction and the OH absorber has been determined from the components of the mode effective charge vector, as defined in Gonze and Lee (1997). The average dielectric susceptibility of the defective crystal was obtained by applying to the defect model the threefold rotation operations consistent with the crystal symmetry. The IR absorption coefficient for polarization perpendicular or parallel to the [001] direction was then calculated from the corresponding diagonal element of the average dielectric tensor.
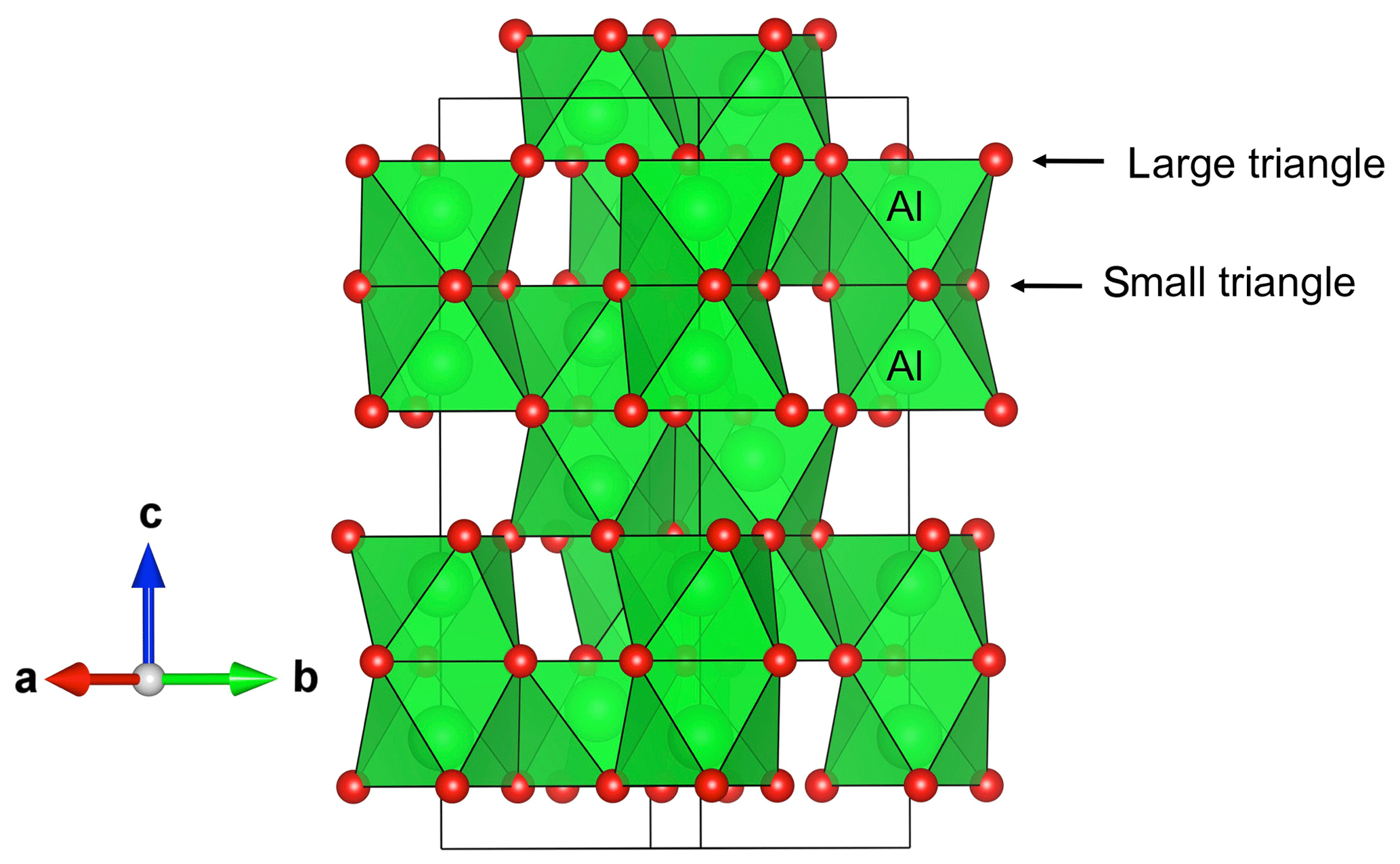
Figure 1View of the corundum structure displaying the pairs of face-sharing octahedral sites. The shared face corresponds to the small oxygen triangle while the opposite face corresponds to the large triangle.
3.1 Hydroxylated intrinsic defects in corundum
The structure of corundum (Newnham and de Haan, 1962) displays pairs of octahedral Al sites parallel to the [001] direction and sharing a triangular face (Fig. 1). The aluminum atoms are not located at the center of the octahedral sites and the Al–O bonds with the three oxygen atoms of the shared face are longer than the three other Al–O bonds. The distance between the O atoms of the shared face is shorter than that observed on the opposite triangular face of the octahedron, forming small and large oxygen triangles, respectively (Fig. 1).
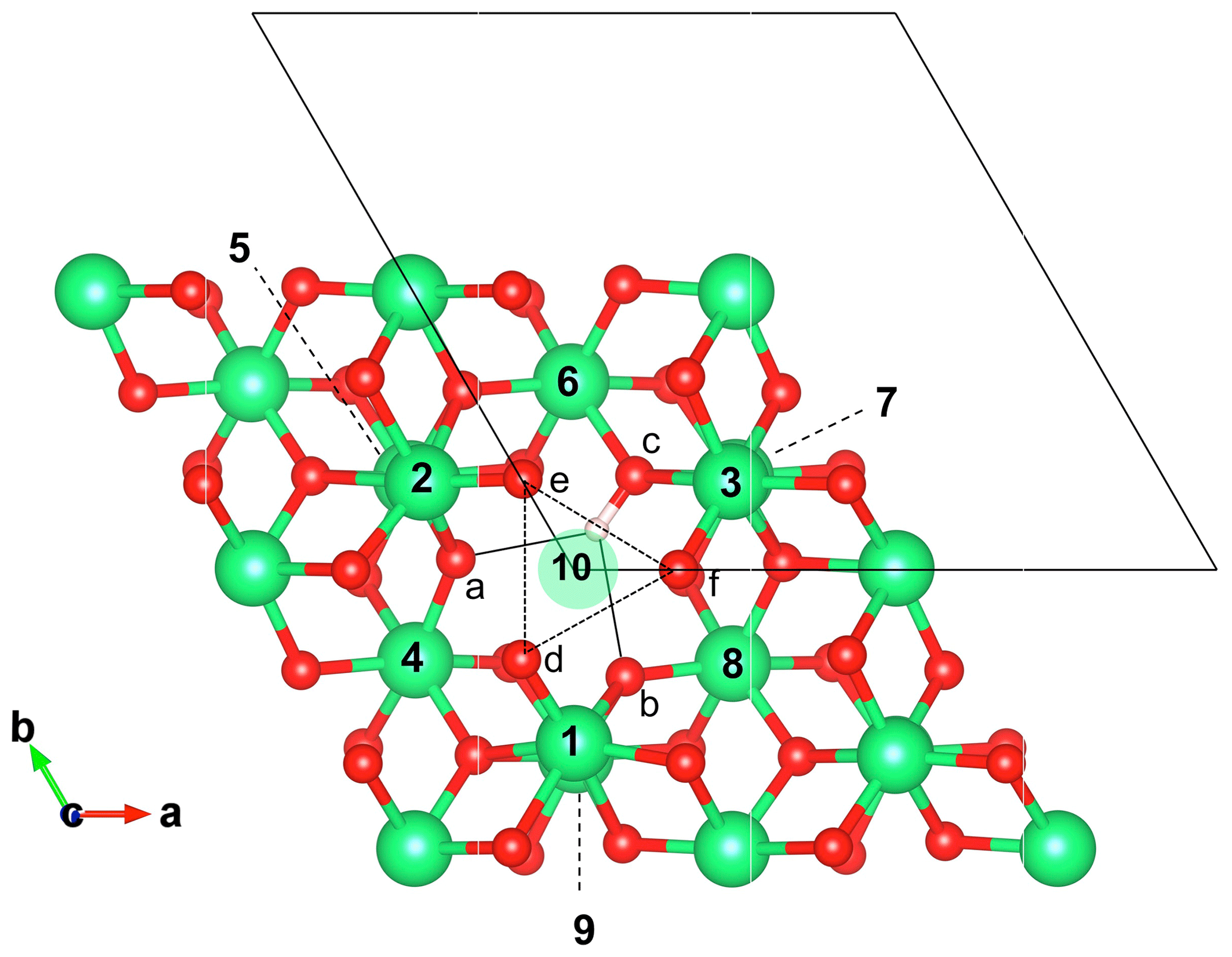
Figure 2Structure of the singly protonated Al vacancy () viewed along the [001] direction. Related models (Table 1) have been obtained by substituting tetravalent cations for Al3+ in nearby sites. The surrounding Al sites are labeled from 1 to 10, and the oxygen sites are labeled from a to f. Note that the Al site 10 corresponds to the octahedral site belonging to the same pair as the vacant site, while sites 1, 2 and 3 belong to the same octahedral layer as the vacant site. The small oxygen triangle (e, f, d) is represented by the dotted lines. The OH group is nearly parallel to the (001) plane and shares a bifurcated hydrogen bond (plain lines) with the facing a and b oxygen atoms. Al, O and H atoms are in green, red and light pink, respectively.
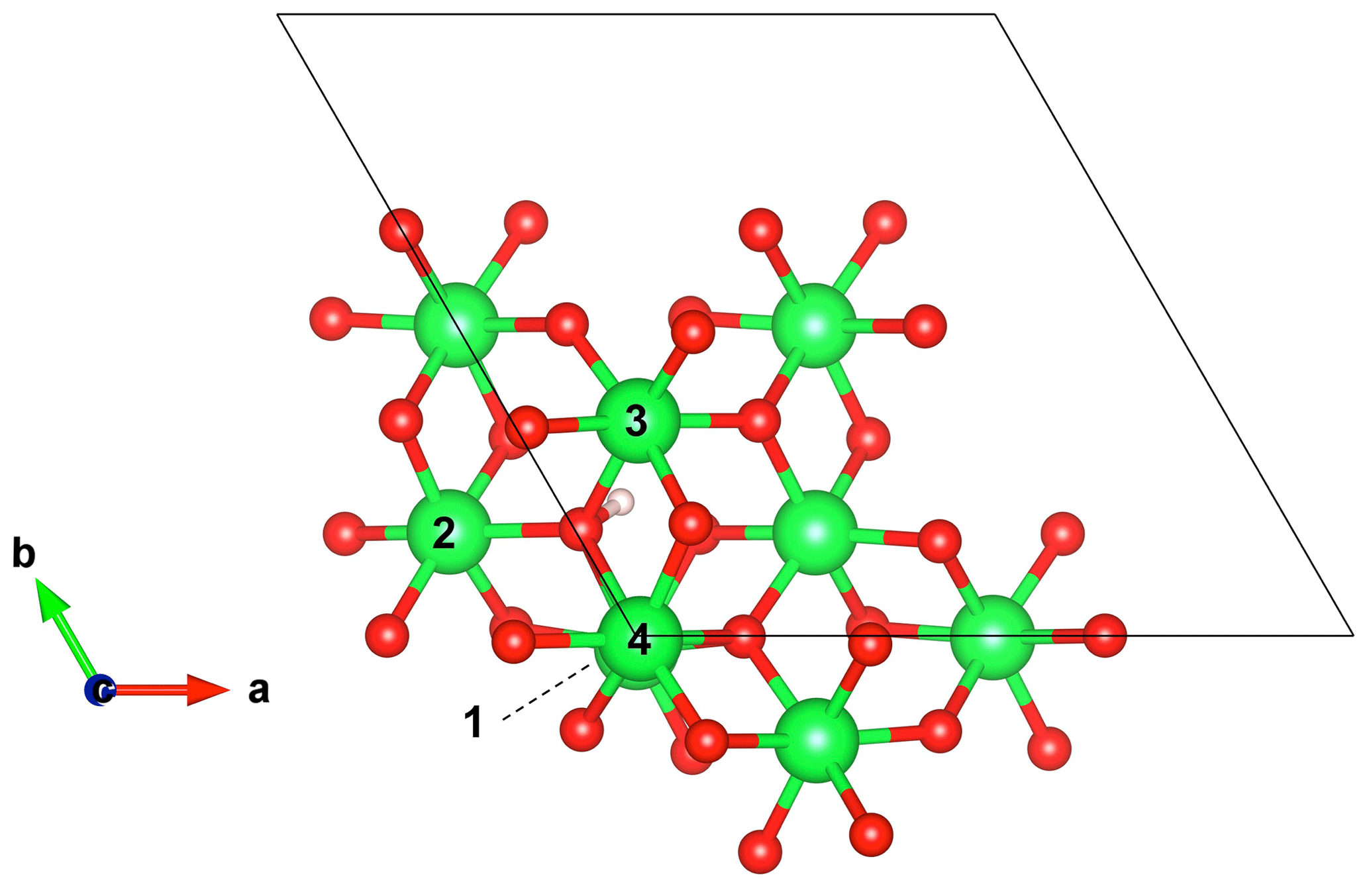
Figure 3Structure of the interstitial proton model ((1H+)i) viewed along the [001] direction. Models of Mg-bearing defects (Table 1) have been obtained by substituting Mg2+ cations for Al3+ in the nearby Al sites labeled from 1 to 4. Note the canting of the OH group with respect to the (001) plane.
Intrinsic defects in corundum involve vacant sites or atoms in interstitial positions (e.g., Lagerlöf and Grimes, 1998). Various schemes of proton association with Al vacancies have previously been investigated by T-Thienprasert et al. (2017). In the present study, models displaying one (1H+)Al or two protons (2H+)Al associated with Al vacancies (Fig. 2), as well as a model of interstitial protons (1H+)i (Fig. 3), have been considered (Table 1). Two (1H+)Al models have been considered depending on the location of the proton on the small or the large oxygen triangle of the vacant site. When the proton is located on an oxygen atom belonging to the large triangle (Fig. 3), the OH group lies almost parallel to the (001) plane and displays a stretching frequency of 3319 cm−1, in excellent agreement with the 3321.67 cm−1 frequency determined by T-Thienprasert et al. (2017). The defect geometry is also consistent with that determined by Zhang et al. (2014). The proton location on an oxygen atom of the small triangle is significantly less favorable, by 50.6 kJ mol−1. In this configuration, the OH group is canted with respect to the (001) plane and displays a lower stretching frequency of 2824.6 cm−1.
Table 1Structural, energetic and vibrational properties of OH defects in corundum (structure files are provided in the Supplement).

* The relative energy is defined with respect to that of the most stable model with the same stoichiometry.
Concerning the doubly protonated Al vacancy, only the configuration displaying two OH groups associated with O atoms belonging to the large triangle has been considered because the geometry of the other configurations determined by T-Thienprasert et al. (2017) are not consistent with the polarization properties of observed OH bands. Under this assumption, this defect has three equivalent configurations due to the threefold symmetry of the site and displays stretching frequencies of 3432.5 and 3378.6 cm−1, consistent with those determined by T-Thienprasert et al. (2017) (model g: 3429 and 3374 cm−1). The geometry (Fig. 3) of the model of the interstitial proton ((1H+)i) is consistent with that previously determined by Zhang et al. (2014) and T-Thienprasert et al. (2017). The theoretical stretching frequency 2994.5 cm−1 is very close to that reported by Thienprasert et al. (2017) (2989.25 cm−1).
A series of electrostatically neutral intrinsic defects involving the association of one proton located in the large oxygen triangle with a combination of Al vacancy and nearby oxygen vacancy has also been examined. The location of the oxygen vacancy in the small oxygen triangle is significantly less favorable (by 50 to 69 kJ mol−1) than in the large triangle, i.e., at a z coordinate similar to that of the OH group (Fig. 2). In the whole series, the OH group still lies nearly parallel to the (001) plane but the presence of the oxygen vacancy affects its length and the related stretching frequency (Table 1).
3.2 Magnesium-associated OH defects in corundum
Defects associating an interstitial proton with a nearby Mg2+ for Al3+ substitution are closely related to the (1H+)i model. Four nonequivalent defect configurations occur depending on the respective position of the OH group and substituted Al site (Fig. 3). These defects are electrostatically neutral, suggesting a greater stability than positively charged interstitial protons. The orientation and length of the OH group are close to those determined for the (1H+)i model, and the corresponding stretching frequencies range between 2990 and 3215 cm−1 (Table 1). Infrared absorption is stronger for a polarization parallel to the [001] direction, corresponding to a theoretical angle between the [001] direction and OH absorber ranging between 32 and 40∘ (Table 2).
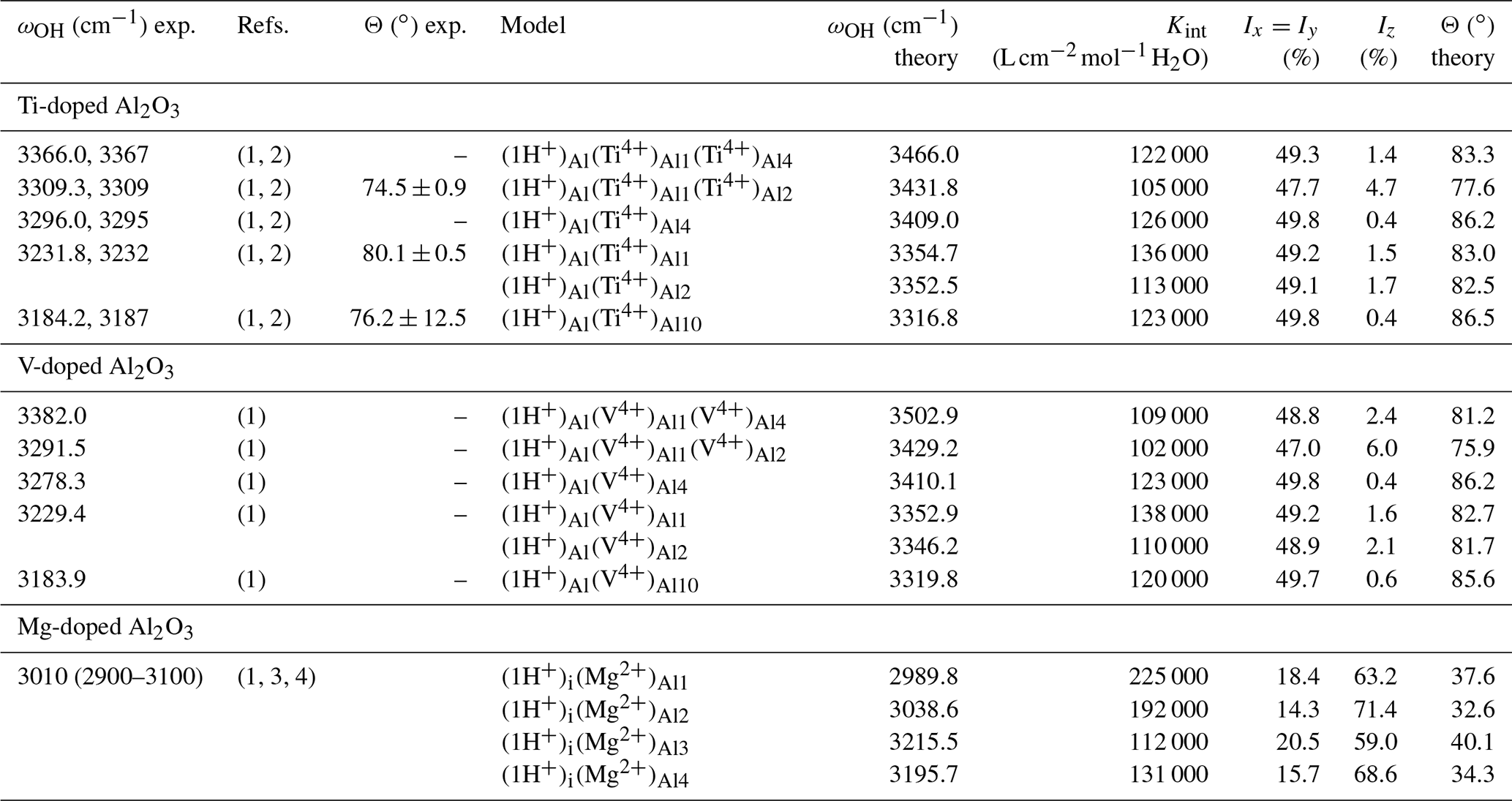
Table 2Experimental and theoretical properties of OH-stretching bands in doped corundum samples. The Ix, Iy and Iz correspond to relative absorption intensities along Cartesian directions, with the z direction being parallel to [001]. The Θ angle between the OH absorber and the [001] direction is determined from the spectroscopic properties.
(1) Ramírez et al. (2004). (2) Moon and Phillips (1991). (3) Volynets et al. (1972). (4) Eigenmann and Günthard (1971).
3.3 Titanium-associated OH defects in corundum
Models of defects associating a single OH group with an Al vacancy and nearby Ti4+ for Al3+ substitutions have been considered. Based on the OH group orientation determined for the intrinsic (1H+)Al models, the OH group is located in the large oxygen triangle of Al vacancy. In this case, its orientation is nearly parallel to the (001) plane, which is consistent with the experimentally observed polarization properties of OH bands in Ti doped samples (Moon and Phillips, 1991, 1994; Ramírez et al., 2004).
Models with one Ti4+ for Al3+ substitution correspond to defects with a −1 electrostatic charge. The 10 configurations (Table 1) correspond to the 10 nonequivalent Al sites closely surrounding the singly protonated Al vacancy (Fig. 2). For these 10 models, the orientation of the OH group remains nearly parallel to the (001) plane. Depending on the respective location of the Ti4+ ion and OH group, the relative stability of the configuration can vary by up to ∼100 kJ mol−1 and the OH stretching frequency displays variations ranging from 3160 to 3410 cm−1. The two most stable configurations correspond to a Ti4+ ion located in a site belonging to the same octahedral layer as the missing Al3+ ion (sites 1 and 2, Fig. 2). Both configurations lead to very close theoretical stretching frequencies at 3354.7 and 3352.5 cm−1. The location of the Ti4+ ion in the site forming an octahedral site pair with the vacant site (site 10, Fig. 2) leads to a configuration with similar stability but displaying a lower stretching frequency (3316.8 cm−1). The bond length and vibrational frequency are similar to those observed for the singly protonated Al vacancy (1H+)Al (3319 cm−1, Table 1). The location of the proton on an oxygen atom belonging to a Ti4+ coordination sphere (corresponding to substitution in sites 3, 6, and 7; Fig. 2) leads to less stable configurations with longer OH bond and lower stretching frequency, consistent with a more significant overbonding of the corresponding oxygen (Table 1).
A second series of models displaying two Ti4+ ions in sites neighboring the Al vacancy has been considered. In this case, the OH-bearing defect is electrostatically neutral. Based on the properties of the singly substituted models, configurations in which the OH group would belong to the coordination sphere of a Ti4+ ion have been discarded, leaving seven potentially substituted sites. The resulting combination of two sites among seven leads to 21 models displaying two Ti4+ ions (Table 1). Consistent with the observations made on singly substituted sites, the most stable configuration is observed for Ti4+ ions located in the two sites at the z coordinate of the missing Al (model ). The corresponding stretching frequency increases to 3431.8 cm−1. The other configurations are less stable, with relative energies varying from 25 to 115 kJ mol−1 and stretching frequencies ranging from 3338 to 3466 cm−1 (Table 1). It is noteworthy that models with one Ti4+ ion located in site 10, i.e., the site forming a pair with the vacant site, display stretching frequencies close to those determined for their counterparts with a single Ti4+ ion close to the Al vacancy. This suggests that infrared spectroscopic data alone might not be sufficient to make a clear-cut distinction between these different Ti clustering patterns.
3.4 Vanadium-associated OH defects in corundum
Infrared spectra of V-doped and Ti-doped corundum samples share some common features in the OH stretching range (Ramírez et al., 2004). Accordingly a series of models containing V4+ ions instead of Ti4+ ions has been considered. This series has been restricted to the most stable configurations obtained for the Ti-associated OH defects. Due to the paramagnetic character of V4+ cations, spin-polarized calculations have been performed, imposing the spin state of isolated ions to the supercell. For systems containing two V4+ cations, the ferromagnetic state has always been found more stable by 0.03 to 5.4 kJ mol−1, depending on the relative position of the V4+ ions. The properties of the defects associated with a single V4+ for Al3+ substitution are very similar to those determined for their Ti bearing counterparts, except moderate variations in the relative energies (Table 1). For the defects associated with two substitutions, the configuration with a pair of V4+ ions facing the OH group displays a higher stretching frequency (3502.9 cm−1) and lower relative energy (14.0 kJ mol−1) than its Ti-bearing counterpart (3466 cm−1 and 33.1 kJ mol−1, respectively).
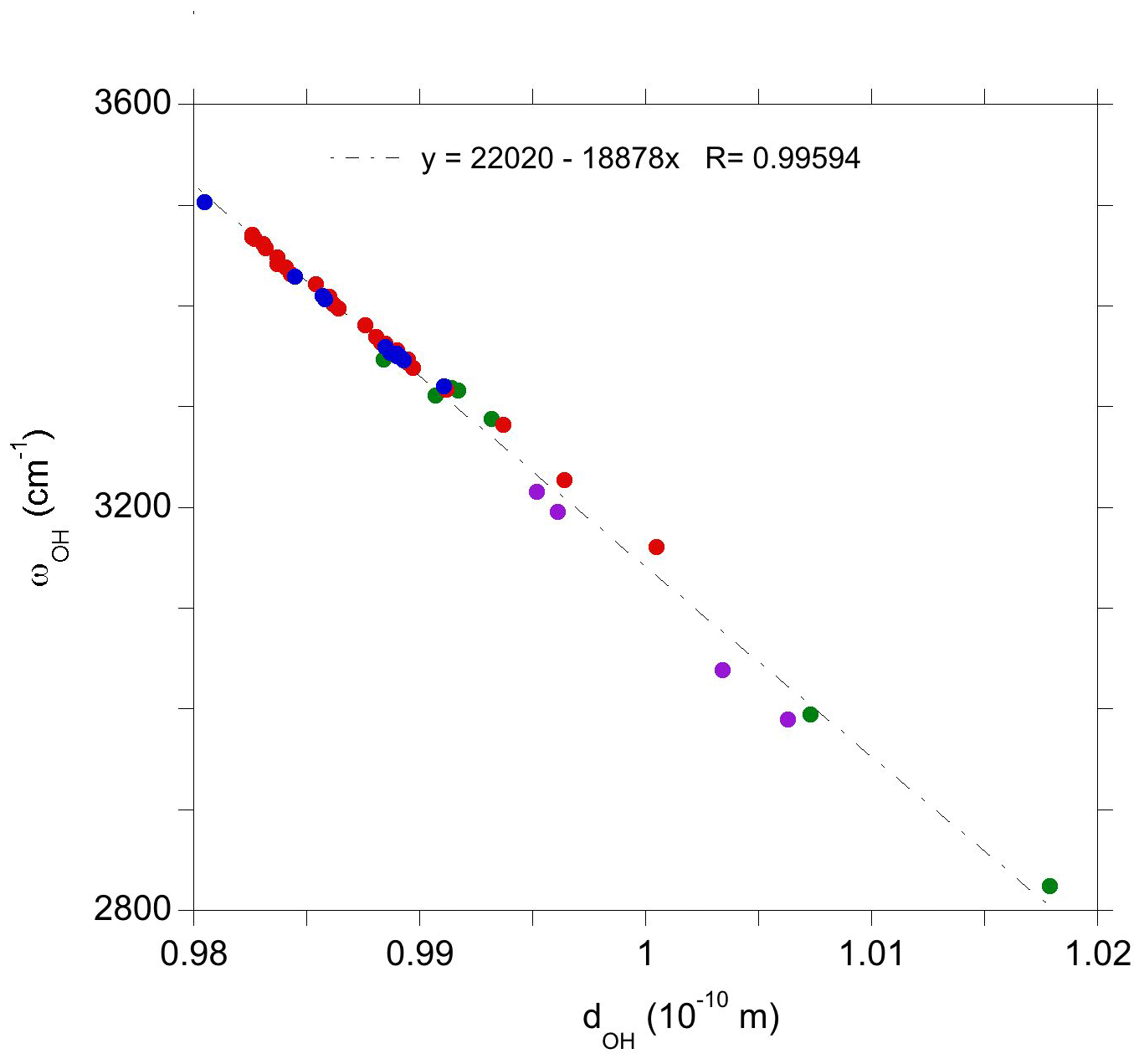
Figure 4Relation between the theoretical stretching frequency and OH bond length for the investigated defects displaying a single OH group. Green: OH associated with intrinsic defects; red: Ti-associated OH defects; blue: V-associated defects; purple: Mg-associated defects.
4.1 General properties of OH defects in corundum
The investigated series of OH defects makes it possible to assess the effect of variations in their atomic-scale environment on their vibrational properties. Discarding the doubly protonated vacancy which displays coupled OH stretching modes, the corresponding series of models display a linear correlation between the stretching frequency and the O–H bond length (Fig. 4). The observed slope (∼19 000 ) is consistent with the slope of 16 000 theoretically determined by Hermansson (1993) for an OH− ion submitted to an external electric field. The observed linear correlation could thus find its origin in the effect of the microscopic electric field acting on the OH group at the defect scale. This is consistent with the frequency increase and OH bond shortening observed in the sequence , and or in its vanadium-bearing counterpart. The presence of one and two substituted tetravalent cations approximately facing the OH group has an increasing contribution to the electric field acting on the OH dipole along a direction parallel to the OH bond. In contrast, the presence of a tetravalent cation in the paired site (site 10, Fig. 2) has a transverse contribution, weakly affecting the microscopic properties of the OH group. This picture is consistent with the interpretation of Ti-associated OH stretching bands using a point charge model by Moon and Phillips (1991, 1994).
It is also noteworthy that the present linear correlation is obtained for a series of defects displaying different charge state (from −2 to +1), which confirms that the addition of a homogeneous electrostatic background to ensure the macroscopic electrostatic neutrality of the periodic system of charged defects does not have a significant effect on the microscopic defect properties, even though the total energy of the system is affected (Leslie and Gillan, 1985; Freysoldt et al., 2009).
4.2 Interpretation of the experimental spectra of doped corundum
Dominant bands in the spectrum of Ti-doped samples are observed at 3309, 3232 and 3184.2 cm−1 (Table 2). The 3309 cm−1 band has been interpreted as being related to a OH group associated with a defect cluster combining an Al vacancy and two nearby Ti4+ cations, while the 3232 and 3184.2 cm−1 bands are only associated with one cation (Moon and Phillips, 1991, 1994). Based on the relative energy and relative stretching frequency of the defect models, the present theoretical results support these previous interpretations. The theoretical polarization properties compare favorably with their experimental counterpart. Theoretical angles between the absorber and the [001] direction are 77.6, 82.5 and 86.5∘ while experimental values determined from polarized infrared spectroscopic measurements are 74.5∘ ± 0.9, 80.1∘ ±0.5 and 76.2∘ ±12, respectively (Moon and Phillips, 1991). The observed relative intensity of the 3232 and 3184.2 cm−1 bands (roughly two-thirds, one-third) in samples fast cooled from 1400 K (Ramírez et al., 2004) is also consistent with a statistical occupancy of three sites with similar stability, two of which lead to close OH-stretching frequencies.
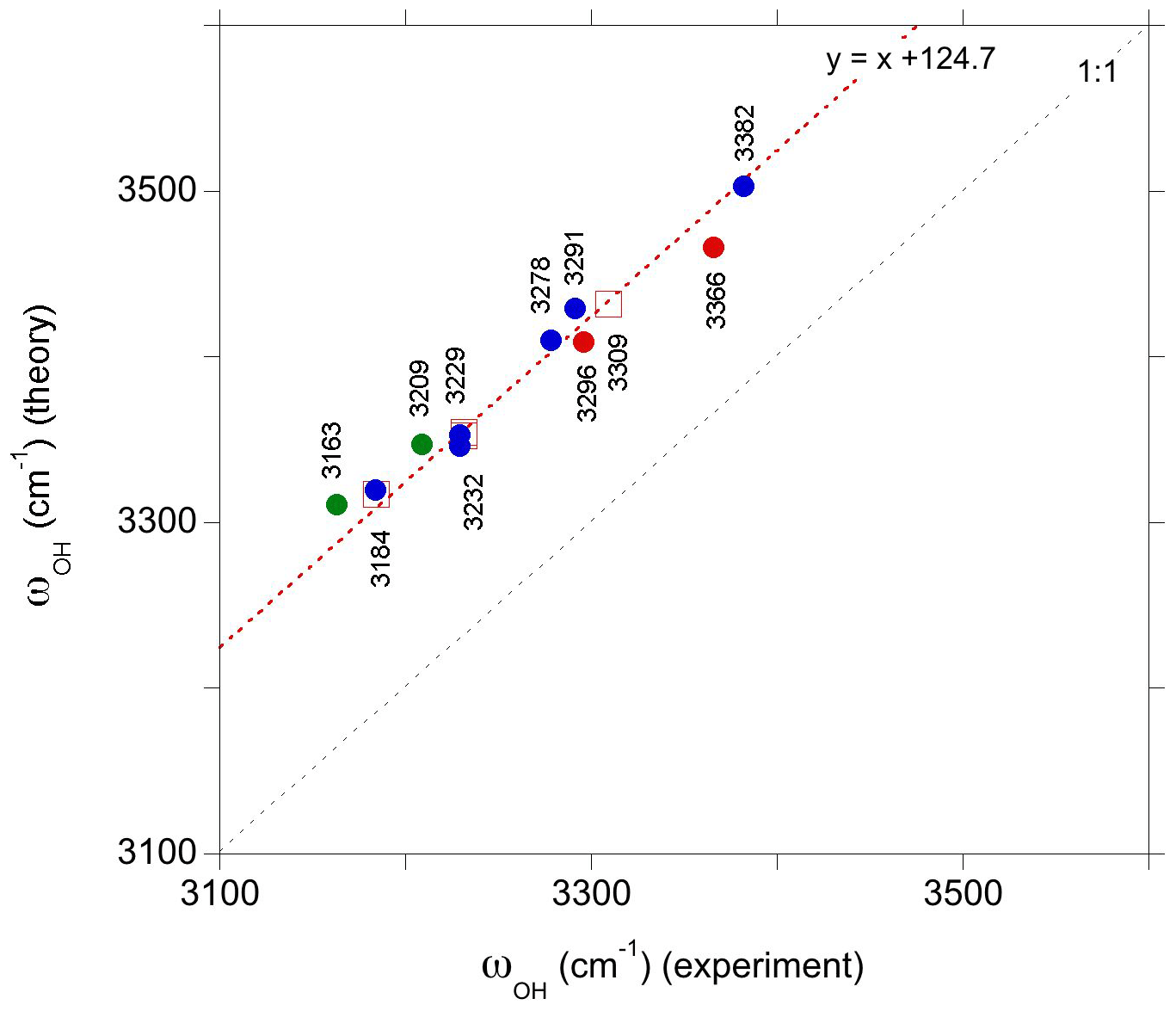
Figure 5Comparison of theoretical and observed frequencies for the relevant defect models. The correlation has been determined using the interpretation proposed for the three major bands observed in Ti-doped samples (open red squares). The other proposed interpretations correspond to the green, red and blue circles for the intrinsic and Ti- and V-bearing defects, respectively. Experimental frequencies are indicated on the figure.
The experimental and theoretical frequencies of these three bands match a linear correlation with a 1:1 slope but reveal a theoretical overestimation of observed frequencies by ∼125 cm−1 (Fig. 5). Similar correlations with a smaller theoretical overestimation of experimental frequencies, ranging from 14 to 50 cm−1, have been observed in previous theoretical studies of OH defects in quartz (Jollands et al., 2020) and diopside (Balan et al., 2020). However, it should be noted that the small difference between the theoretical and observed frequencies mostly results from a systematic cancelation between the use of GGA and the neglect of anharmonicity in the determination of theoretical frequencies (Balan et al., 2007; Finnochi et al., 2008). Experimental harmonic frequencies are typically more than 160 cm−1 higher than their anharmonic counterparts in moderately H-bonded systems (e.g., Szalay et al., 2002). Therefore, a difference between theoretical harmonic and experimentally observed frequencies amounting to 125 cm−1 is still reasonable.
Based on the interpretation of the bands related to Ti-associated OH defects (Table 2), their counterparts in the V-doped sample of Ramírez et al. (2004) can be assigned to the corresponding V-associated OH defects. The band at 3291.5 cm−1 is therefore ascribed to an Al vacancy associated with two V4+ cations (model ), while the bands at 3229.4 and 3183.9 cm−1 correspond to configurations with a single V4+ ion. It is noteworthy that these two bands are observed at very similar frequencies in Ti- or V-doped samples. Thus, it does not seem possible to discriminate the related Ti-associated and V-associated OH defects in samples containing both trace elements. In contrast, the bands at 3309 and 3291.5 cm−1 can be observed in synthetic “alexandrite”-type samples containing V and Ti impurities (Beran, 1991).
The bands observed at 3366 and 3382 cm−1 in the Ti-doped and V-doped samples, respectively, match the comparatively higher stretching frequencies determined for the and models, still assuming a ∼125 cm−1 shift between theoretical and experimental frequencies (Fig. 5). In these models the tetravalent cations occupy a pair of edge-sharing sites facing the OH group (Fig. 2). This tight configuration is consistent with an increased contribution of the tetravalent cations to the electric field affecting the OH dipole and increasing its stretching frequency. Based on stretching frequencies, it is also possible to propose models with a M4+ for Al3+ substitution in site 4 to interpret the two bands observed at 3296 and 3278.3 cm−1 in Ti-doped and V-doped samples, respectively.
An additional difficulty in the interpretation of OH stretching bands observed in Ti- or V-doped corundum arises due to the weak effect on the OH properties of the presence of a tetravalent cation in the octahedral site forming a face-sharing pair with the vacant site (site 10, Fig. 2). Thus, a contribution of configurations displaying two tetravalent cations cannot be excluded in the interpretation of bands observed at frequencies lower than 3300 cm−1. Additional constraints can be obtained using the relative energy determined for the various configurations of a given defect stoichiometry. In a Boltzmann formalism, the probability of a single configuration of relative energy Ei among a total of N non-degenerated configurations is assessed by the expression
where T is the temperature and R the ideal gas constant. Resulting configuration distributions have been calculated for temperatures of 900 and 1400 K and for the defects associated with one or two Ti4+ or V4+ cations (Table 1; Fig. 6). The 1400 K temperature corresponds to the hydrogenation temperature used by Ramírez et al. (2004) while the 900 K temperature is slightly below that of the lowest isochronal heating experiments (973 K) of Moon and Phillips (1991, 1994).
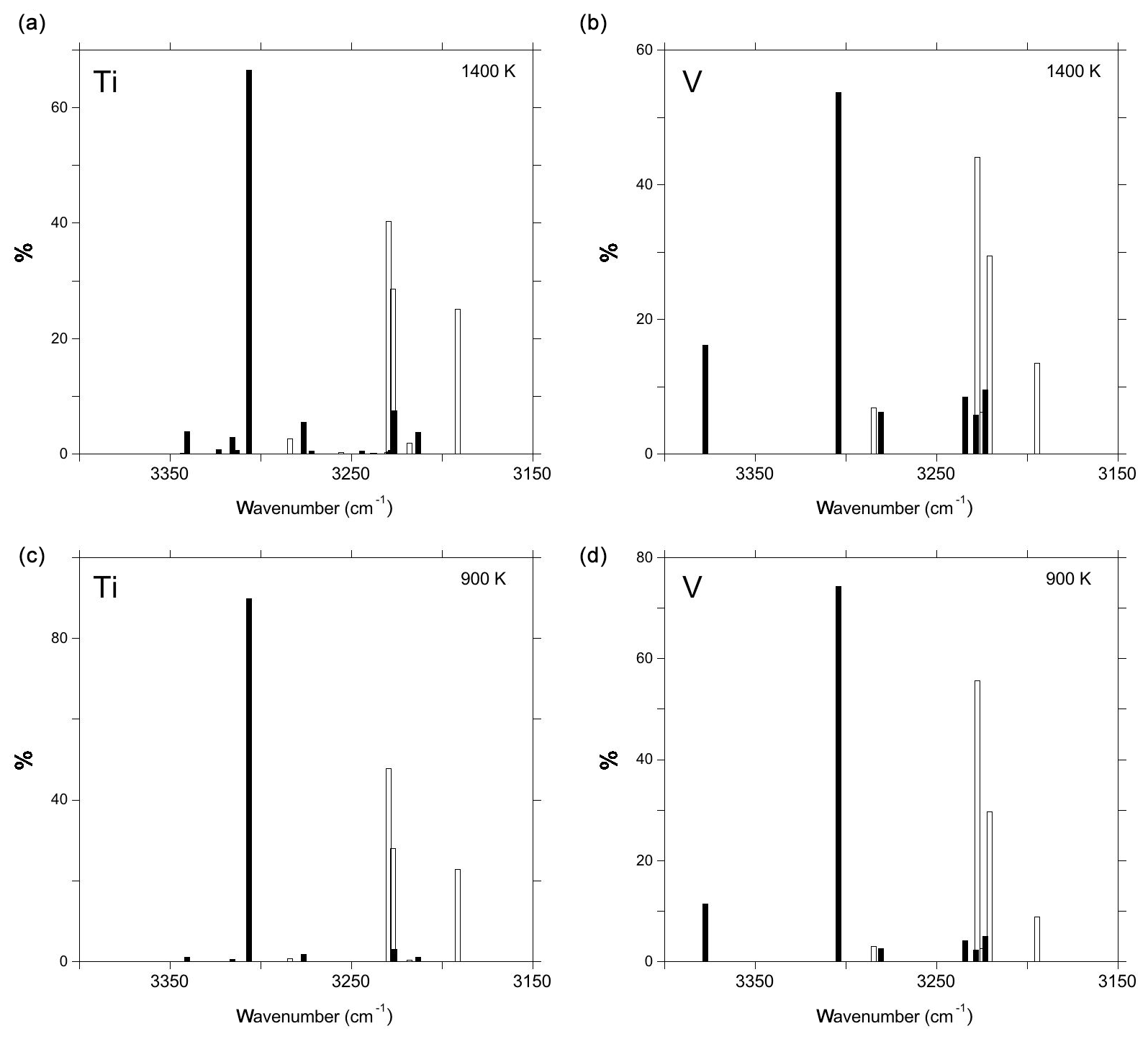
Figure 6Configuration distributions of Ti-associated defects (a, c) and V-associated defects (b, d) at temperatures of 900 K (a, b) and 1400 K (c, d). The open and full bars correspond to defect models displaying one or two cationic substitutions, respectively. The theoretical frequencies have been lowered by 125 cm−1 to facilitate the comparison with experimental spectra. Note that the clustering equilibrium describing the relative contributions of models displaying one or two cationic substitutions is not considered in these graphs.
The absorption coefficients computed for the relevant defect models display moderate variations from 100 000 to 140 000 (Table 2). These values are close to those inferred from the experimental calibration of Libowitzky and Rossman (1997) for band frequencies around 3300 cm−1. Accordingly, the intensity variations observed in the experimental spectra as a function of the treatment temperature or cooling rate of the sample are dominated by variations in the clustering pattern of tetravalent cations, with more clustered configurations favored at lower temperatures (Moon and Phillips, 1991, 1994). Comparing the spectra of fast-cooled samples of Ramírez et al. (2004) with the configuration distributions computed at 1400 K, it can be inferred that the configuration responsible for at 3309 cm−1 band dominates the population of defects with two Ti4+ cations. Accordingly, a significant contribution of defects associated with two cations to the other bands at lower frequency is unlikely. By implication, this supports the attribution of the band at 3296 cm−1 to the defect. Similar reasoning supports the attribution of the band at 3278.3 cm−1 to the defect. Overall, the configuration distributions are consistent with the relative intensity variations observed between the spectra of fast-cooled and slow-cooled Ti- and V-doped samples reported by Ramírez et al. (2004), suggesting that these spectra correspond to defect populations prevailing at higher and lower temperatures, respectively.
Although the band observed at 3010 cm−1 in the Mg-doped samples (Volynets et al., 1972; Ramírez et al., 1997b) is significantly broader than those observed in Ti- or V-doped samples, the models of interstitial protons associated with substituted Mg2+ ions are consistent with experimental observation in terms of polarization properties and frequencies (Table 2). The theoretical Iz∕Ix ratio of the models (Table 2) ranges between 2.88 and 4.99, which compares favorably with the 3.33 ratio reported by Volynets et al. (1972) and with the 5:1 ratio reported for the similar bands observed in Ni- or Co-doped samples (Eigenmann and Günthard, 1971). The four defect models lead to two stretching frequencies close to 3000 cm−1 and two higher frequencies close to 3200 cm−1, which could explain the band splitting observed in the slow-cooled Mg-doped sample of Ramírez et al. (2004).
4.3 Interpretation of the experimental spectra of synthetic nominally pure corundum
Comparing with doped samples, the interpretation of spectra recorded on nominally pure samples is not so straightforward. The band observed at 3278 cm−1 by Turner and Crawford (1975) and Engstrom et al. (1980) in UV-grade corundum does not seem to match any of the intrinsic defects investigated in the present study, while its frequency perfectly coincides with that observed for the defect in V-doped samples. However, the lack of bands related to other defect configurations in the spectrum reported by Turner and Crawford (1975) challenges its assignment to an Al vacancy with cationic substitutions distributed over neighboring sites. In the spectra of the U and H samples of Ramírez et al. (2004), and in the hydrothermally treated sample of Kronenberg et al. (2000), the 3278.3 cm−1 band is associated with bands at 3231.4 and 3183.7 cm−1, but here again the relative intensities of the bands, as well as their thermally induced variations, differ from those expected for an equilibrium distribution of defects, as inferred for the doped samples. On another side, the polarization properties and stretching frequency of this band are more consistent with an association with an Al vacancy, as proposed by Turner and Crawford (1975), than with a location on the small oxygen triangle separating the paired Al sites as proposed by Engstrom et al. (1980). In addition, the angle of 86∘ between the OH absorber and the [001] direction determined by Ramírez et al. (2004) compares very well with the theoretical value (86.2∘) determined for the model (Table 2). It is also striking that some of the new bands in the spectrum of a nominally pure sample exposed to UV irradiation display frequencies similar to those observed in Ti- or V-doped samples (Ramírez et al., 2004). Although some unidentified defects might fortuitously lead to same OH orientations and stretching frequencies, it is also possible that the observed bands still correspond to the impurity-associated OH defects identified in doped samples but displaying a far-from-equilibrium distribution of configurations. Of note, only non-paramagnetic intrinsic defects have been considered in the present study. While they did not find any evidence for the paramagnetic center proposed by Turner and Crawford (1975), Engstrom et al. (1980) did not exclude interaction of hydrogen atoms with an unidentified impurity to explain the 3279 cm−1 band. The oxidation of traces of structural Ti3+ or V3+ ions during sample treatments may also lead to a more complex behavior than that expected from a simple configuration equilibrium.
Finally, the weaker 3163 and 3209 cm−1 bands observed by Kronenberg et al. (2000) in a hydrothermally treated synthetic sample could be tentatively related to the (1H+)AlVOb and (1H+)AlVOa defects, respectively (Table 1; Fig. 5). These two OH defects of similar stability correspond to an electrostatically neutral association with nearby Al and oxygen vacancies (Table 1). The 3160 cm−1 band has also been reported by Beran (1991) in Verneuil-grown corundum samples. It can also be noted that according to their theoretical frequencies, the intrinsic defects could also overlap with the band at 3183 cm−1. It is however expected that intrinsic defects only play a minor role in hydrogen incorporation in corundum, due to the strong reduction of their concentration by dopant ions, even when these ions occur at trace impurity levels (Lagerlöf and Grimes, 1998).
Based on the comparison of the theoretical spectroscopic properties of defect models with experimental data, the observed OH stretching bands in Ti- or V-doped corundum have been related to specific atomic-scale configurations of OH defects. The OH groups are dominantly associated with clustered defects formed by cationic vacancies and neighboring tetravalent impurities. The models are consistent with those previously proposed by Moon and Phillips (1991, 1994), even though some details of the atomic-scale geometry may differ. Most likely, they also hold for natural samples, which always contain significant impurity concentrations (e.g., Muhlmeister et al., 1998) and display spectroscopic features similar to those of the doped synthetic samples (e.g., Beran, 1991; Smith, 1995; Beran and Rossman, 2006).
Chemically complex Ti-bearing defects have previously been proposed as playing a role in the hydrogen speciation in olivine (Berry et al., 2005; Tollan et al., 2017) and diopside (Balan et al., 2020). While these defects could have a global role when observed in major phases of the Earth mantle (e.g., Faul et al., 2016; Demouchy and Bolfan Casanova, 2016), their presence and configurational equilibrium in natural samples could also provide information on the physical–chemical conditions and timescales of geological processes (Padrón-Navarta et al., 2014). In the case of corundum, the identified OH defects could have relevant applications for the determination of the sources and potential heat treatments of rubies and sapphires (Smith, 1995).
Structure drawings have been produced with the Vesta software (Momma and Izumi, 2011). PWscf and PHonon codes (Giannozzi et al., 2009) are available at http://www.quantum-espresso.org/ (last access: 8 September 2020). Defect structures are provided in the Supplement under “.vesta” and “.cif” formats.
The supplement related to this article is available online at: https://doi.org/10.5194/ejm-32-457-2020-supplement.
The author declares that there is no conflict of interest.
Reviews by Dmitry Krylov and the one anonymous reviewer are acknowledged with appreciation. This work was granted access to the HPC resources of IDRIS under the allocation 2020-A0080910820 attributed by GENCI (Grand Equipement National de Calcul Intensif).
This paper was edited by Roland Stalder and reviewed by Dmitry Krylov and one anonymous referee.
Balan, E., Lazzeri, M., Delattre, S., Meheut, M., Refson, K., and Winkler, B.: Anharmonicity of inner-OH stretching modes in hydrous phyllosilicates: assessment from first-principles frozen-phonon calculations, Phys. Chem. Miner., 34, 621–625, 2007.
Balan, E., Refson, K., Blanchard, M., Delattre, S., Lazzeri, M., Ingrin, J., Mauri, F., Wright, K., and Winkler, B.: Theoretical infrared absorption coefficient of OH groups in minerals, Am. Mineral., 93, 950–953, https://doi.org/10.2138/am.2008.2889, 2008.
Balan, E., Blanchard, M., Lazzeri, M., and Ingrin, J.: Theoretical Raman spectrum and anharmonicity of tetrahedral OH defects in hydrous forsterite, Eur. J. Mineral., 29, 201–212, https://doi.org/10.1127/ejm/2017/0029-2599, 2017.
Balan, E., Paulatto, L., Liu, J., and Ingrin, J.: Low-temperature infrared spectrum and atomic-scale structure of hydrous defects in diopside, Eur. J. Mineral., submitted, 2020.
Baroni, S., de Gironcoli, S., Dal Corso, A., and Giannozzi, P.: Phonons and related crystal properties from density-functional perturbation theory, Rev. Mod. Physics, 73, 515–561, https://doi.org/10.1103/RevModPhys.73.515, 2001.
Beran, A.: Trace hydrogen in Verneuil-grown corundum and its colour varieties – an IR pectroscopic study, Eur. J. Mineral., 3, 971–975, 1991.
Beran, A. and Rossman, G. R.: OH in naturally occurring corundum, Eur. J. Mineral., 18, 441–447, 2006.
Berry, A. J., Hermann, J., O'Neill, H. S. C., and Foran, G. J.: Finger- printing the water site in mantle olivine, Geology, 33, 869–872, https://doi.org/10.1130/G21759.1, 2005.
Blanchard, M., Ingrin, J., Balan, E., Kovács, I., and Withers, A. C.: Effect of iron and trivalent cations on OH-defects in olivine, Am. Mineral., 102, 302–311, https://doi.org/10.2138/am-2017-5777, 2017.
Bowles, J. F. W., Howie, R. A., Vaughan, D. J., and Zussman, J.: Rock-forming Minerals. Non-silicates: Oxides, Hydroxides and Sulphides, 5A, 920 pp., Geological Society, London, 2011.
Demouchy, S. and Bolfan-Casanova, N.: Distribution and transport of hydrogen in the lithospheric mantle: A review, Lithos, 240–243, 402–425, 2016.
Eigenmann, K. and Günthard, H. H.: Hydrogen incorporation in doped α-Al2O3 by high temperature redox reactions, Chem. Phys. Lett., 12, 12–15, 1971.
Engstrom, H., Bates, J. B., Wang, J. C., and Abraham, M. M.: Infrared spectra of hydrogen isotopes in α-Al2O3, Phys. Rev. B, 21, 1520–1526, 1980.
Faul, U. H., Cline, C. J., David, E. C., Berry, A. J., and Jackson, I.: Titanium-hydroxyl defect- controlled rheology of the Earth's upper mantle, Earth. Planet. Sci. Lett., 452, 227–237, 2016.
Finnochi, F., Hacquart, R., Naud, C., and Jupille J.: Hydroxyl-defect complexes on hydrated MgO smokes, J. Phys. Chem. C, 112, 13226–13231, 2008.
Freysoldt, C., Neugebauer, J., and Van de Walle, C.: Fully ab initio finite-size corrections for charged-defect supercell calculations, Phys. Rev. Lett., 102, 016402, https://doi.org/10.1103/PhysRevLett.102.016402, 2009.
Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Chiarotti, G. L., Cococcioni, M., Dabo, I., Dal Corso, A., de Gironcoli, S., Fabris, S., Fratesi, G., Gebauer, R., Gerstmann, U., Gougoussis, C., Kokalj, A., Lazzeri, M., Martin-Samos, L., Marzari, N., Mauri, F., Mazzarello, R., Paolini, S., Pasquarello, A., Paulatto, L., Sbraccia, C., Scandolo, S., Sclauzero, G., Seitsonen, A. P., Smogunov, A., Umari, P., and Wentzcovitch, R. M.: Quantum ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.-Cond. Mat., 21, 395502, https://doi.org/10.1088/0953-8984/21/39/395502, 2009.
Gonze, X., and Lee, C.: Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional pertubation theory, Phys. Rev. B, 55, 10355–10368, 1997.
Hamann, D. R.: Optimized norm-conserving Vanderbilt pseudopotentials, Phys. Rev. B, 88, 085117, https://doi.org/10.1103/PhysRevB.88.085117, 2013.
Hermansson, K.: Redshifts and blueshifts of OH vibrations, Int. Jour. Quantum. Chem., 45, 747–758, 1993.
Jollands, M. C., Blanchard, M., and Balan, E.: Structure and infrared spectra of OH-defects in quartz, Eur. J. Mineral., 32, 311–323, 2020.
Kronenberg, A. K., Castaing, J., Mitchell, T. E., and Kirby, S. H.: Hydrogen defects in α-Al2O3 and water weakening of sapphire and alumina ceramics between 600 and 1000 ∘C – I. Infrared characterization of defects, Acta Mater., 48, 1481–1494, 2000.
Lagerlöf, K. P. D. and Grimes, R. W.: The defect chemistry of sapphire (α-Al2O3), Acta Mater. 46, 5689–5700, 1998.
Leslie, M. and Gillan, M. J.: The energy and elastic dipole tensor of defects in ionic crystals calculated by the supercell method, J. Phys. Chem. 18, 973–982, 1985.
Libowitzky, E. and Beran, A.: The structure of hydrous species in nominally anhydrous minerals: Information from polarized IR spectroscopy, Rev. Mineral. Geochem., 62, 29–52, 2006.
Libowitzky, E. and Rossman, G. R.: An IR absorption calibration for water in minerals, Am. Mineral., 82, 1111–1115, 1997.
Momma, K. and Izumi, F.: VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 44, 1272–1276, https://doi.org/10.1107/S0021889811038970, 2011.
Moon, A. R. and Phillips, M. R.: Defect clustering in H,Ti: α-Al2O3, J. Phys. Chem. Solids., 52, 1087–1099, 1991.
Moon, A. R. and Phillips, M. R.: Defect clustering and color in Fe,Ti: α-Al2O3, J. Amer. Ceram. Soc., 77, 356–367, 1994.
Muhlmeister, S., Fritsch, E., Shigley, J. E., Devouard, B., and Laurs, B. M.: Separating natural and synthetic rubies on the basis of trace-element chemistry, Gems and Gemmology, 34, 80-101, 1998.
Newnham, R. E. and de Haan, Y. M.: Refinement of the alpha Al2O3, Ti2O3, V2O3 and Cr2O3 structures, Zeit. Kristall., 117, 235–237, 1962.
Padrón-Navarta, J. A., Hermann, J., and O'Neill, H. S. C.: Site-specific hydrogen diffusion rates in forsterite, Earth Planet. Sc. Lett., 392, 100–112, 2014.
Perdew, J. P., Burke, K., and Ernzerhof, M.: Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 77, 3865–3868, https://doi.org/10.1103/PhysRevLett.77.3865, 1996.
Ramírez, R., González, R., Colera, I., and Chen, Y.: Electric-field-enhanced diffusion of deuterons and protons in α-Al2O3 crystals, Phys. Rev. B, 55, 237–242, 1997a.
Ramírez, R., González, R., Colera, I., and Vila, R.: Protons and deuterons in magnesium-doped sapphire crystals, J. Am. Ceram. Soc., 80, 847–850, 1997b.
Ramírez, R., Colera, I., González, R., Chen, Y., and Kokta, M. R.: Hydrogen-isotope transport induced by an electric field in α-Al2O3 single crystals, Phys. Rev. B, 69, 014302, https://doi.org/10.1103/PhysRevB.69.014302, 2004.
Rossman, G. R.: The geochemistry of gems and its relevance to gemology: Different traces, different prices, Elements, 5, 159–162, https://doi.org/10.2113/gselements.5.3.159, 2009.
Schlipf, M. and Gygi, F.: Optimization algorithm for the generation of ONCV pseudopotentials, Comput. Phys. Comm., 196, 36, https://doi.org/10.1016/j.cpc.2015.05.011, 2015.
Smith, C. P.: A contribution to understanding the infrared spectra of rubies from Mong Hsu, Myanmar, J. Gemm., 24, 321–335, 1995.
Szalay, V., Kovács, L., Wöhlecke, M., and Libowitzky, E.: Stretching potential and equilibrium length of the OH bonds in solids, Chem. Phys. Lett., 354, 56–61, 2002.
T-Thienprasert, J., Boonchun, A., Reunchan, P., and Limpijumnong, S.: Identification of hydrogen defects in α-Al2O3 by first-principles local vibration mode calculations, Phys. Rev. B, 95, 134103, https://doi.org/10.1103/PhysRevB.95.134103, 2017.
Tollan, P. M. E., Smith, R., St. C. O'Neill, H., and Hermann, J.: The responses of the four main substitution mechanisms of H in olivine to H2O activity at 1050 ∘C and 3 GPa, Prog. Earth Planet. Sci., 4, 14, https://doi.org/10.1186/s40645-017-0128-7, 2017.
Turner, T. J. and Crawford Jr., J. H.: V centers in single crystal Al2O3, Solid State Comm., 17 167–169, 1975.
Volynets, F. K., Sidorova, Y. A., and Stsepuro, N. A.: OH-groups in corundum crystals grown by the Verneuil technique, J. Appl. Spectr., 17, 1088–1091, 1972.
Wöhlecke, M. and Kovaács, L.: OH ions in oxide crystal, Critical Reviews in Solid State and Material Sciences, 25, 1–86, 2001.
Wright, K.: Atomistic models of OH defects in nominally anhydrous minerals, Rev. Mineral. Geochem., 62, 67–83, 2006.
Zhang, G., Lu, Y., and Wang, X.: Hydrogen interactions with intrinsic point defects in hydrogen permeation barrier of α-Al2O3: a first-principles study, Phys. Chem. Chem. Phys., 16, 17523, https://doi.org/10.1039/c4cp01382d, 2014.